Inflammatory myopathy
Inflammatory myopathy is disease featuring weakness and inflammation of muscles and (in some types) muscle pain. The cause of much inflammatory myopathy is unknown (idiopathic), and such cases are classified according to their symptoms and signs and electromyography, MRI and laboratory findings. It can also be associated with underlying cancer. The main classes of idiopathic inflammatory myopathy are polymyositis (PM), dermatomyositis (DM), and inclusion-body myositis (IBM).
| Inflammatory myopathy | |
|---|---|
| Other names | Inflammatory muscle disease or Myositis |
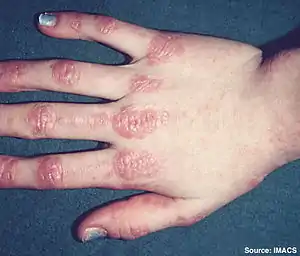 | |
| Gottron's papules on the hand of a patient with juvenile dermatomyositis. | |
| Specialty | Rheumatology |
Diagnosis
There are a number of known causes of myopathy, and it is only once these have been ruled out that a clinician will assign an idiopathic inflammatory myopathy (IIM) syndrome to a case. The usual criteria for a diagnosis of PM are weakness in muscles of the head, neck, trunk, upper arms or upper legs; raised blood serum concentrations of some muscle enzymes such as creatine kinase; unhealthy muscle changes on electromyography; and biopsy findings of (i) muscle cell degeneration and regeneration and (ii) chronic inflammatory infiltrates in muscle cells. If heliotrope (purple) rash or Gottron's papules are also present, then the diagnosis is DM. In DM, myositis may not be clinically apparent but detectable via biopsy or MRI.[1] If the criteria for PM are met but muscle weakness also affects the hands and feet or is not accompanied by pain IBM should be suspected, and confirmed when muscle cell biopsy reveals (i) cytoplasmic vacuoles fringed by basophilic granules and (ii) inflammatory infiltrate comprising mostly CD8 T lymphocytes and macrophages; and electron microscopy reveals filamentous inclusions in both cytoplasm and nucleus.[2]
Treatment
There have been few randomized treatment trials, due to the relative rarity of inflammatory myopathies.[3] The goal of treatment is improvement in activities of daily living and muscle strength. Suppression of immune system activity (immunosuppression) is the treatment strategy. Patients with PM or DM almost always improve to some degree in response to treatment, at least initially, and many recover fully with maintenance therapy. (If there is no initial improvement from treatment of PM or DM, the diagnosis should be carefully re-examined.) There is no proven effective therapy for IBM, and most IBM patients will need assistive devices such as a cane, a walking frame or a wheelchair. The later in life IBM arises, the more aggressive it appears to be.[4]
Polymyositis and dermatomyositis
In severe cases of PM and DM with systemic signs, an initial three to five days on intravenous corticosteroid (methylprednisolone) may be used; but normally treatment begins with a single daily (after breakfast) high dose of oral corticosteroid (prednisone). After a month or so the strength of every second day's dose is very gradually reduced over three to four months, to minimize the negative effects of the prednisone. When a high dose of prednisone cannot be reduced without losing muscle strength, or when prednisone is effective but it is producing significant complications, "steroid sparing" oral immunosuppressants such as azathioprine, mycophenolate mofetil, methotrexate and cyclosporine, may be used in combination with reduced prednisone. Some of these steroid sparing drugs can take several months to demonstrate an effect.[4]
To minimize side effects, patients on corticosteroids should follow a strict high-protein, low-carbohydrate, low-salt diet; and with long-term corticosteroid use a daily calcium supplement and weekly vitamin D supplement (and a weekly dose of Fosamax for postmenopausal women) should be considered.[4]
For patients not responding to this approach there is weak evidence supporting the use of intravenous immunoglobulin, ciclosporin, tacrolimus, mycophenolate mofetil and other agents; and trials of rituximab have indicated a potential therapeutic effect.[3]
Inclusion-body myositis
Despite its very similar clinical presentation to PM, IBM does not respond to the drugs that effectively treat PM, and there is no proven effective therapy for IBM.[4] Alemtuzumab is being studied but as of May 2013 it had not demonstrated clinical effectiveness in IBM.[3] Dysphagia (difficulty swallowing) may be improved by intravenous immunoglobulin, though more trials are needed.[5] Non-fatiguing, systematic strength-building exercise has demonstrated benefit.[6] Occupational and rehabilitation therapists can offer good advice on walking without falling and performing fine motor tasks, and can provide appropriate canes, braces and wheelchairs. Speech pathologists can provide advice on preventing choking episodes and reducing the anxiety of an immanent aspiration for both patients and carers.[4]
Epidemiology
Every year between 2.18 and 7.7 people per million receive a diagnosis of PM or DM.[7] Around 3.2 children per million per year are diagnosed with DM (termed juvenile dermatomyositis), with an average age of onset of seven years. Diagnosis of adult DM commonly occurs between 30 and 50 years of age. PM is an adult disease, usually emerging after the age of twenty. PM and DM are more common in females, more common in Caucasians, and least common in Asians. At any given time, about 35.5 people per million have IBM; it emerges after the age of 30 (usually after 50), and may be more common in males.[3]
References
- Miller, Frederick W (10 June 2009). "Classification of Idiopathic Inflammatory Myopathies". In Kagen, Lawrence J (ed.). The Inflammatory Myopathies. Springer. pp. 23–36. ISBN 9781627038393.
- Kagen, Lawrence J (10 June 2009). "Inclusion Body Myositis". The Inflammatory Myopathies. Springer. pp. 95–102. ISBN 9781627038393.
- Holton, Janice L; Wedderburn, Lucy R; Hanna, Michael G (29 May 2013). "Polymyositis, Dermatomyositis and Inclusion Body Myositis". In Goebel, Hans H; Sewry, Caroline A; Weller, Roy O (eds.). Muscle Disease: Pathology and Genetics. John Wiley & Sons. pp. 747–781. ISBN 978-1-118-63549-0.
- Dalakas, Marinos C (8 September 2010). "Treatment and Management of Autoimmune Myopathies". In Bertorini, Tulio E (ed.). Neuromuscular Disorders. Elsevier Health Sciences. pp. 395–408. ISBN 9781437703726.
- Cited in Dalakas, Marinos C (08 September 2010). "Treatment and Management of Autoimmune Myopathies". In Bertorini, Tulio E. Neuromuscular Disorders. Elsevier Health Sciences. pp. 395—408. ISBN 9781437703726:
- Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K (March 1997). "Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study". Neurology. 48 (3): 712–6. doi:10.1212/wnl.48.3.712. PMID 9065553.
- Walter MC, Lochmüller H, Toepfer M, et al. (January 2000). "High-dose immunoglobulin therapy in sporadic inclusion body myositis: a double-blind, placebo-controlled study". J. Neurol. 247 (1): 22–8. doi:10.1007/s004150050005. PMID 10701893.
- Cherin P, Pelletier S, Teixeira A, et al. (January 2002). "Intravenous immunoglobulin for dysphagia of inclusion body myositis". Neurology. 58 (2): 326. doi:10.1212/wnl.58.2.326. PMID 11805271.
- Spector SA, Lemmer JT, Koffman BM, et al. (October 1997). "Safety and efficacy of strength training in patients with sporadic inclusion body myositis". Muscle Nerve. 20 (10): 1242–8. doi:10.1002/(sici)1097-4598(199710)20:10<1242::aid-mus6>3.3.co;2-x. PMID 9324080. Cited in:
- Dalakas, Marinos C (08 September 2010). "Treatment and Management of Autoimmune Myopathies". In Bertorini, Tulio E. Neuromuscular Disorders. Elsevier Health Sciences. pp. 395—408. ISBN 9781437703726
- Mastaglia FL, Garlepp MJ, Phillips BA, Zilko PJ (April 2003). "Inflammatory myopathies: clinical, diagnostic and therapeutic aspects". Muscle Nerve. 27 (4): 407–25. doi:10.1002/mus.10313. PMID 12661042. Cited in:
- Holton, Janice L; Wedderburn, Lucy R; Hanna, Michael G (29 May 2013). "Polymyositis, Dermatomyositis and Inclusion Body Myositis". In Goebel, Hans H; Sewry, Caroline A; Weller, Roy O (eds.). Muscle Disease: Pathology and Genetics. John Wiley & Sons. pp. 747–781. ISBN 978-1-118-63549-0.