Kikuchi disease
Kikuchi disease was described in 1972 in Japan. It is also known as histiocytic necrotizing lymphadenitis, Kikuchi necrotizing lymphadenitis, phagocytic necrotizing lymphadenitis, subacute necrotizing lymphadenitis, and necrotizing lymphadenitis.[1][2][3][4] Kikuchi disease occurs sporadically in people with no family history of the condition.[5]
| Kikuchi disease | |
|---|---|
| Other names | Histiocytic necrotizing lymphadenitis, Kikuchi-Fujimoto disease |
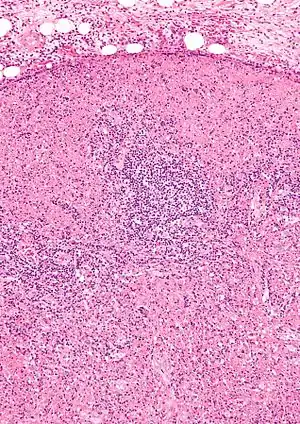 | |
| Micrograph of a lymph node with Kikuchi disease showing the characteristic features (abundant histiocytes, necrosis without neutrophils). H&E stain. | |
| Specialty | Angiology |
It was first described by Dr Masahiro Kikuchi (1935–2012) in 1972[6] and independently by Y. Fujimoto.
Signs and symptoms
The signs and symptoms of Kikuchi disease are fever, enlargement of the lymph nodes (lymphadenopathy), skin rashes, and headache.[7] Rarely, enlargement of the liver and spleen and nervous system involvement resembling meningitis are seen. Often a bout of extreme fatigue can occur - often taking hold during latter parts of the day and the affected person can be more prone to fatigue from exercise.
Pathophysiology
Some studies have suggested a genetic predisposition to the proposed autoimmune response. Several infectious candidates have been associated with Kikuchi disease.[8]
Many theories exist about the cause of KFD. Microbial/viral or autoimmune causes have been suggested. Mycobacterium szulgai and Yersinia and Toxoplasma species have been implicated. More recently, growing evidence suggests a role for Epstein-Barr virus, as well as other viruses (HHV6, HHV8, parvovirus B19, HIV and HTLV-1) in the pathogenesis of KFD.[1] However, many independent studies have failed to identify the presence of these infectious agents in cases of Kikuchi lymphadenopathy.[9] In addition, serologic tests including antibodies to a host of viruses have consistently proven noncontributory and no viral particles have been identified ultrastructurally. KFD is now proposed to be a nonspecific hyperimmune reaction to a variety of infectious, chemical, physical, and neoplastic agents. Other autoimmune conditions and manifestations such as antiphospholipid syndrome, polymyositis, systemic juvenile idiopathic arthritis, bilateral uveitis, arthritis and cutaneous necrotizing vasculitis have been linked to KFD. KFD may represent an exuberant T-cell-mediated immune response in a genetically susceptible individual to a variety of nonspecific stimuli.[1]
Certain Human leukocyte antigen class II genes appear more frequently in patients with Kikuchi disease, suggesting that there may be a genetic predisposition to the proposed autoimmune response.[10]
Diagnosis
It is diagnosed by lymph node excision biopsy. Kikuchi disease is a self-limiting illness which has symptoms which may overlap with Hodgkin's lymphoma leading to misdiagnosis in some patients. Antinuclear antibodies, antiphospholipid antibodies, anti-dsDNA, and rheumatoid factor are usually negative, and may help in differentiation from systemic lupus erythematosus.[11]
Differential diagnosis
The differential diagnosis of Kikuchi disease includes systemic lupus erythematosus (SLE), disseminated tuberculosis, lymphoma, sarcoidosis, and viral lymphadenitis. Clinical findings sometimes may include positive results for IgM/IgG/IgA antibodies. For other causes of lymph node enlargement, see lymphadenopathy.[12]
Management
No specific cure is known. Treatment is largely supportive. Nonsteroidal anti-inflammatory drugs (NSAIDs) are indicated for tender lymph nodes and fever, and corticosteroids are useful in severe extranodal or generalized disease.[13]
Symptomatic measures aimed at relieving the distressing local and systemic complaints have been described as the main line of management of KFD. Analgesics, antipyretics, NSAIDs, and corticosteroids have been used. If the clinical course is more severe, with multiple flares of bulky enlarged cervical lymph nodes and fever, then a low-dose corticosteroid treatment has been suggested.[14]
Epidemiology
Kikuchi-Fujimoto disease (KFD) is a rare, self-limiting disorder that typically affects the cervical lymph nodes. Recognition of this condition is crucial, especially because it can easily be mistaken for tuberculosis, lymphoma, or even adenocarcinoma. Awareness of this disorder helps prevent misdiagnosis and inappropriate treatment.[1]
Kikuchi's disease is a very rare disease mainly seen in Japan. Isolated cases are reported in North America, Europe, Asia, England, and at least two cases in New Zealand. It is possible that the prevalence of KFD is greater than is reported given lymphadenopathy can be overlooked and the disease's self-limiting nature. That a definite identification of KFD can only be done via a biopsy of affected tissues further suggests that cases go unrecognized or undiagnosed. [15] It is mainly a disease of young adults (20–30 years), with a slight bias towards females. The cause of this disease is not known, although infectious and autoimmune causes have been proposed. The course of the disease is generally benign and self-limiting. Lymph node enlargement usually resolves over several weeks to six months. The recurrence rate is about 3%. Death from Kikuchi disease is extremely rare and usually occurs due to liver, respiratory, or heart failure.
See also
References
- Rammohan A, Cherukuri SD, Manimaran AB, Manohar RR, Naidu RM (June 2012). "Kikuchi-Fujimoto Disease: A Sheep in Wolf's Clothing". J Otolaryngol Head Neck Surg. 41 (3): 222–26. PMID 22762705.
- Kaushik V, Malik TH, Bishop PW, Jones PH (June 2004). "Histiocytic necrotising lymphadenitis (Kikuchi's disease): a rare cause of cervical lymphadenopathy". Surgeon. 2 (3): 179–82. doi:10.1016/s1479-666x(04)80084-2. PMID 15570824.
- Bosch X, Guilabert A (2006). "Kikuchi-Fujimoto disease". Orphanet J Rare Dis. 1: 18. doi:10.1186/1750-1172-1-18. PMC 1481509. PMID 16722618.
- Bosch X, Guilabert A, Miquel R, Campo E (July 2004). "Enigmatic Kikuchi-Fujimoto disease: a comprehensive review". Am. J. Clin. Pathol. 122 (1): 141–52. doi:10.1309/YF08-1L4T-KYWV-YVPQ. PMID 15272543.
- "kikuchi disease". Retrieved 17 April 2018.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytes. Acta Hematol Jpn 1972;35:379–80.
- "Kikuchi disease". Genetic and Rare Diseases Information Center (GARD). Retrieved 17 April 2018.
- Atwater AR, Longley BJ, Aughenbaugh WD (July 2008). "Kikuchi's disease: case report and systematic review of cutaneous and histopathologic presentations". J. Am. Acad. Dermatol. 59 (1): 130–36. doi:10.1016/j.jaad.2008.03.012. PMID 18462833.
- Rosado, F. G.; Tang, Y. W.; Hasserjian, R. P.; McClain, C. M.; Wang, B; Mosse, C. A. (2013). "Kikuchi-Fujimoto lymphadenitis: Role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8". Human Pathology. 44 (2): 255–59. doi:10.1016/j.humpath.2012.05.016. PMID 22939574.
- Tanaka, T.; Ohmori, M.; Yasunaga, S.; Ohshima, K.; Kikuchi, M.; Sasazuki, T. (1999). "DNA typing of HLA class II genes (HLA-DR, -DQ and -DP) in Japanese patients with histiocytic necrotizing lymphadentis (Kikuchi's disease) 8". Tissue Antigens. 54 (3): 246–253. doi:10.1034/j.1399-0039.1999.540305.x. PMID 10519361.
- Bosch X, Guilabert A (23 May 2006). "Kikuchi-Fujimoto disease". Orphanet Journal of Rare Diseases. 1 (18): 18. doi:10.1186/1750-1172-1-18. PMC 1481509. PMID 16722618.
- "Lymphadenopathy". The Lecturio Medical Concept Library. Retrieved 7 August 2021.
- Bosch X, Guilabert A (23 May 2006). "Kikuchi-Fujimoto disease". Orphanet Journal of Rare Diseases. 1 (18): 18. doi:10.1186/1750-1172-1-18. PMC 1481509. PMID 16722618.
- Bosch X, Guilabert A (23 May 2006). "Kikuchi-Fujimoto disease". Orphanet Journal of Rare Diseases. 1 (18): 18. doi:10.1186/1750-1172-1-18. PMC 1481509. PMID 16722618.
- Ulug, Mehmet; Aslan, Vahap (March 1, 2012). "An unusual cause of cervical lymphadenopathy: Kikuchi-Fujimoto disease". Journal of Microbiology and Infectious Diseases. 2 (1): 21–25. doi:10.5799/ahinjs.02.2012.01.0036.