Kuru (disease)
Kuru is a rare, incurable, and fatal neurodegenerative disorder that was formerly common among the Fore people of Papua New Guinea. Kuru is a form of transmissible spongiform encephalopathy (TSE) caused by the transmission of abnormally folded proteins (prions), which leads to symptoms such as tremors and loss of coordination from neurodegeneration.
| Kuru | |
|---|---|
 | |
| A Fore child with advanced kuru. He is unable to walk or sit upright without assistance and is severely malnourished. | |
| Specialty | Neuropathology, infectious disease |
| Symptoms | Body tremors, random outbursts of laughter, gradual loss of coordination |
| Complications | Infection and pneumonia during the terminal stage. |
| Usual onset | Often takes years or even decades for symptoms to appear after exposure |
| Duration | 11–14 month life expectancy after onset of symptoms[1] |
| Causes | Transmission of infected prion proteins |
| Risk factors | Cannibalism |
| Diagnostic method | Autopsy |
| Differential diagnosis | Creutzfeldt–Jakob disease |
| Prevention | Avoid practices of cannibalism |
| Treatment | None |
| Prognosis | Always fatal |
| Frequency | 2,700 (1957–2004) |
| Deaths | Approximately 2,700 |
The term kuru derives from the Fore word kuria or guria ("to shake"),[2] due to the body tremors that are a classic symptom of the disease. Kúru itself means "trembling".[3] It is also known as the "laughing sickness" due to the pathologic bursts of laughter which are a symptom of the disease. It is now widely accepted that kuru was transmitted among members of the Fore tribe of Papua New Guinea via funerary cannibalism. Deceased family members were traditionally cooked and eaten, which was thought to help free the spirit of the dead.[4] Women and children usually consumed the brain, the organ in which infectious prions were most concentrated, thus allowing for transmission of kuru. The disease was therefore more prevalent among women and children.
The epidemic likely started when a villager developed sporadic Creutzfeldt–Jakob disease and died. When villagers ate the brain, they contracted the disease and then spread it to other villagers who ate their infected brains.[5]
While the Fore people stopped consuming human meat in the early 1960s, when it was first speculated to be transmitted via endocannibalism, the disease lingered due to kuru's long incubation period of anywhere from 10 to over 50 years.[6] The epidemic finally declined sharply after half a century, from 200 deaths per year in 1957 to no deaths from at least 2010 onwards, with sources disagreeing on whether the last known kuru victim died in 2005 or 2009.[7][8][9][10]
Signs and symptoms
Kuru, a transmissible spongiform encephalopathy, is a disease of the nervous system that causes physiological and neurological effects which ultimately lead to death. It is characterized by progressive cerebellar ataxia, or loss of coordination and control over muscle movements.[11][12]
The preclinical or asymptomatic phase, also called the incubation period, averages 10–13 years, but can be as short as five and has been estimated to last as long as 50 years or more after initial exposure.[13]
The clinical stage, which begins at the first onset of symptoms, lasts an average of 12 months. The clinical progression of kuru is divided into three specific stages: the ambulant, sedentary and terminal stages. While there is some variation in these stages between individuals, they are highly conserved among the affected population.[11] Before the onset of clinical symptoms, an individual can also present with prodromal symptoms including headache and joint pain in the legs.[14]
In the first (ambulant) stage, the infected individual may exhibit unsteady stance and gait, decreased muscle control, tremors, difficulty pronouncing words (dysarthria), and tremors titubation. This stage is named the ambulant because the individual is still able to walk around despite symptoms.[14]
In the second (sedentary) stage, the infected individual is incapable of walking without support and experiences ataxia and severe tremors. Furthermore, the individual shows signs of emotional instability and depression, yet exhibits uncontrolled and sporadic laughter. Despite the other neurological symptoms, tendon reflexes are still intact at this stage of the disease.[14]
In the third and final (terminal) stage, the infected individual's existing symptoms, like ataxia, progress to the point where it is no longer possible to sit up without support. New symptoms also emerge: the individual develops dysphagia, which can lead to severe malnutrition, and may also become incontinent, lose the ability or will to speak, and become unresponsive to their surroundings despite maintaining consciousness.[14] Towards the end of the terminal stage, patients often develop chronic ulcerated wounds that can be easily infected. An infected person usually dies within three months to two years after the first terminal stage symptoms, often because of pneumonia or other secondary infections.[15]
Causes
Kuru is largely localized to the Fore people and people with whom they intermarried.[16] The Fore people ritualistically cooked and consumed body parts of their family members following their death to incorporate "the body of the dead person into the bodies of living relatives, thus helping to free the spirit of the dead".[17] Because the brain is the organ enriched in the infectious prion, women and children, who consumed brain, had a much higher likelihood of being infected than men, who preferentially consumed muscles.[18]
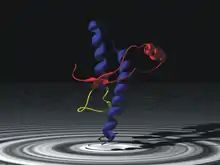
Prion

The infectious agent is a misfolded form of a host-encoded protein called prion (PrP). Prion proteins are encoded by the Prion Protein Gene (PRNP).[20] The two forms of prion are designated as PrPc, which is a normally folded protein, and PrPsc, a misfolded form which gives rise to the disease. The two forms do not differ in their amino acid sequence; however, the pathogenic PrPsc isoform differs from the normal PrPc form in its secondary and tertiary structure. The PrPsc isoform is more enriched in beta sheets, while the normal PrPc form is enriched in alpha helices.[18] The differences in conformation allow PrPsc to aggregate and be extremely resistant to protein degradation by enzymes or by other chemical and physical means. The normal form, on the other hand, is susceptible to complete proteolysis and soluble in non-denaturing detergents.[14]
It has been suggested that pre-existing or acquired PrPsc can promote the conversion of PrPc into PrPsc, which goes on to convert other PrPc. This initiates a chain reaction that allows for its rapid propagation, resulting in the pathogenesis of prion diseases.[14]
Transmission
In 1961, Australian medical researcher Michael Alpers conducted extensive field studies among the Fore accompanied by anthropologist Shirley Lindenbaum.[9] Their historical research suggested the epidemic may have originated around 1900 from a single individual who lived on the edge of Fore territory and who is thought to have spontaneously developed some form of Creutzfeldt–Jakob disease.[21] Alpers and Lindenbaum's research conclusively demonstrated that kuru spread easily and rapidly in the Fore people due to their endocannibalistic funeral practices, in which relatives consumed the bodies of the dead to return the person's "life force" to the hamlet, a Fore societal subunit.[22] Corpses of family members were often buried for days, then exhumed once the corpses were colonized by insect larvae, at which point the corpse would be dismembered and served with the larvae as a side dish.[23]
The demographic distribution evident in the infection rates – kuru was eight to nine times more prevalent in women and children than in men at its peak – is because Fore men considered consuming human flesh to weaken them in times of conflict or battle, while the women and children were more likely to eat the bodies of the deceased, including the brain, where the prion particles were particularly concentrated. Also, the strong possibility exists that it was passed on to women and children more easily because they took on the task of cleaning relatives after death and might have had open sores and cuts on their hands.[18]
Although ingestion of the prion particles can lead to the disease,[24] a high degree of transmission occurred if the prion particles could reach the subcutaneous tissue. With elimination of cannibalism because of Australian colonial law enforcement and the local Christian missionaries' efforts, Alpers' research showed that kuru was already declining among the Fore by the mid‑1960s. However, the mean incubation period of the disease is 14 years, and 7 cases were reported with latencies of 40 years or more for those who were most genetically resilient, continuing to appear for several more decades. Sources disagree on whether the last person with kuru died in 2005 or 2009.[9][10][7][8]
Immunity
In 2009, researchers at the Medical Research Council discovered a naturally occurring variant of a prion protein in a population from Papua New Guinea that confers strong resistance to kuru. In the study, which began in 1996,[25] researchers assessed over 3,000 people from the affected and surrounding Eastern Highland populations, and identified a variation in the prion protein G127.[26] G127 polymorphism is the result of a missense mutation, and is highly geographically restricted to regions where the kuru epidemic was the most widespread. Researchers believe that the PrnP variant occurred very recently, estimating that the most recent common ancestor lived 10 generations ago.[26]
The findings of the study could help researchers better understand and develop treatments for other related prion diseases, such as Creutzfeldt–Jakob disease[25] and Alzheimer's disease.[27]
History
Kuru was first described in official reports by Australian officers patrolling the Eastern Highlands of Papua New Guinea in the early 1950s.[28] Some unofficial accounts place kuru in the region as early as 1910.[7] In 1951, Arthur Carey was the first to use the term kuru in a report to describe a new disease afflicting the Fore tribes of Papua New Guinea (PNG). In his report, Carey noted that kuru mostly affected Fore women, eventually killing them. Kuru was noted in the Fore, Yate and Usurufa people in 1952-1953 by anthropologists Ronald Berndt and Catherine Berndt.[7] In 1953, kuru was observed by patrol officer John McArthur, who provided a description of the disease in his report. McArthur believed that kuru was merely a psychosomatic episode resulting from the sorcery practices of the tribal people in the region.[28] After the disease had progressed into a larger epidemic, the tribal people asked Charles Pfarr, a Lutheran medical officer, to come to the area to report the disease to Australian authorities.[7]
Initially, the Fore people believed the causes of kuru to be sorcery or witchcraft.[29] They also thought that the magic causing kuru was contagious. It was also called negi-nagi, which meant foolish person as the victims laughed at spontaneous intervals.[30] This disease, the Fore people believed, was caused by ghosts, because of the shaking and strange behaviour that comes with kuru. Attempting to cure this, they would feed victims casuarina bark.[31]
When kuru disease had become an epidemic, Daniel Carleton Gajdusek, a virologist, and Vincent Zigas, a medical doctor, started research on the disease. In 1957, Zigas and Gajdusek published a report in the Medical Journal of Australia that suggested that kuru had a genetic origin, and that "any ethnic-environmental variables that are operating in kuru pathogenesis have not yet been determined."[32]
Cannibalism was suspected as a possible cause from the very beginning but was not formally put forth as a hypothesis until 1967 by Glasse and more formally in 1968 by Mathews, Glasse, and Lindenbaum.[30]
Even before anthropophagy had been linked to kuru, cannibalism was banned by the Australian administration of Papua New Guinea, and the practice was nearly eliminated by 1960. While the number of cases of kuru was decreasing, medical researchers were finally able to properly investigate kuru, which eventually led to the modern understanding of prions as its cause.[33]
In an effort to understand the pathology of kuru disease, Gajdusek established the first experimental tests on chimpanzees for kuru at the National Institutes of Health (NIH).[7] Michael Alpers, an Australian doctor, collaborated with Gajdusek by providing samples of brain tissues he had taken from an 11-year-old Fore girl who had died of kuru. In his work, Gajdusek was also the first to compile a bibliography of kuru disease.[34] Joe Gibbs joined Gajdusek to monitor and record the behavior of the apes at the NIH and conduct their autopsies. Within two years, one of the chimps, Daisy, had developed kuru, demonstrating that an unknown disease factor was transmitted through infected biomaterial and that it was capable of crossing the species barrier to other primates. After Elisabeth Beck confirmed that this experiment had brought about the first experimental transmission of kuru, the finding was deemed a very important advance in human medicine, leading to the award of the Nobel Prize in Physiology or Medicine to Gajdusek in 1976.[7]
Subsequently, E. J. Field spent large parts of the late 1960s and early 1970s in New Guinea investigating the disease,[35] connecting it to scrapie and multiple sclerosis.[36] He noted the disease's interactions with glial cells, including the critical observation that the infectious process may depend on the structural rearrangement of the host's molecules.[37] This was an early observation of what was to later become the prion hypothesis.[38]
See also
- Cannibalism
- Donner Party
- Endocannibalism
- Exocannibalism
- List of incidents of cannibalism
- Transmissible spongiform encephalopathy
References
- "The epidemiology of kuru in the period 1987 to 1995", Department of Health (Australia), retrieved February 5, 2019
- Hoskin, J.O.; Kiloh, L.G.; Cawte, J.E. (April 1969). "Epilepsy and guria: The shaking syndromes of New Guinea". Social Science & Medicine. 3 (1): 39–48. doi:10.1016/0037-7856(69)90037-7. PMID 5809623.
- Scott, Graham (1978). The Fore Language of Papua New Guinea. Pacific Linguistics. pp. 2, 6. doi:10.15144/PL-B47. hdl:1885/146489. ISBN 978-0-85883-173-5.
- Whitfield, Jerome T; Pako, Wandagi H; Collinge, John; Alpers, Michael P (27 November 2008). "Mortuary rites of the South Fore and kuru". Philosophical Transactions of the Royal Society B: Biological Sciences. 363 (1510): 3721–3724. doi:10.1098/rstb.2008.0074. PMC 2581657. PMID 18849288.
- Bichell, Rae Ellen (September 6, 2016). "When People Ate People, A Strange Disease Emerged". NPR.org. Retrieved 2018-04-08.
- "Kuru". MedlinePlus Medical Encyclopedia. Retrieved 2016-11-14.
- Alpers, MP (2007). "A history of kuru". Papua and New Guinea Medical Journal. 50 (1–2): 10–9. PMID 19354007.
- Rense, Sarah (September 7, 2016). "Here's What Happens to Your Body When You Eat Human Meat". Esquire.
- "A life of determination". Monash University — Faculty of Medicine, Nursing and Health Sciences. 2009-02-23. Archived from the original on 2015-12-10. Retrieved 20 January 2016.
- Collinge, John; Whitfield, Jerome; McKintosh, Edward; Beck, John; Mead, Simon; Thomas, Dafydd J; Alpers, Michael P (June 2006). "Kuru in the 21st century—an acquired human prion disease with very long incubation periods". The Lancet. 367 (9528): 2068–2074. doi:10.1016/S0140-6736(06)68930-7. PMID 16798390. S2CID 11506094.
- Alpers, Michael P (December 2005). "The epidemiology of kuru in the period 1987 to 1995". Communicable Diseases Intelligence. 29 (4): 391–399. PMID 16465931. Retrieved 2016-11-10.
- Liberski, Pawel P.; Sikorska, Beata; Lindenbaum, Shirley; Goldfarb, Lev G.; McLean, Catriona; Hainfellner, Johannes A.; Brown, Paul (February 2012). "Kuru". Journal of Neuropathology & Experimental Neurology. 71 (2): 92–103. doi:10.1097/NEN.0b013e3182444efd. PMC 5120877. PMID 22249461.
- Collinge, John; Whitfield, Jerome; McKintosh, Edward; Frosh, Adam; Mead, Simon; Hill, Andrew F; Brandner, Sebastian; Thomas, Dafydd; Alpers, Michael P (27 November 2008). "A clinical study of kuru patients with long incubation periods at the end of the epidemic in Papua New Guinea". Philosophical Transactions of the Royal Society B: Biological Sciences. 363 (1510): 3725–3739. doi:10.1098/rstb.2008.0068. PMC 2581654. PMID 18849289.
- Imran, Muhammad; Mahmood, Saqib (24 December 2011). "An overview of human prion diseases". Virology Journal. 8 (1): 559. doi:10.1186/1743-422X-8-559. PMC 3296552. PMID 22196171.
- Wadsworth, Jonathan D. F.; Joiner, Susan; Linehan, Jacqueline M.; Desbruslais, Melanie; Fox, Katie; Cooper, Sharon; Cronier, Sabrina; Asante, Emmanuel A.; Mead, Simon; Brandner, Sebastian; Hill, Andrew F.; Collinge, John (11 March 2008). "Kuru prions and sporadic Creutzfeldt–Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice". Proceedings of the National Academy of Sciences of the United States of America. 105 (10): 3885–3890. Bibcode:2008PNAS..105.3885W. doi:10.1073/pnas.0800190105. PMC 2268835. PMID 18316717.
- Lindenbaum, Shirley (2001-01-01). "Kuru, Prions, and Human Affairs: Thinking About Epidemics". Annual Review of Anthropology. 30 (1): 363–385. doi:10.1146/annurev.anthro.30.1.363. S2CID 162196301.
- Whitfield, Jerome T.; Pako, Wandagi H.; Collinge, John; Alpers, Michael P. (2008-11-27). "Mortuary rites of the South Fore and kuru". Philosophical Transactions of the Royal Society B: Biological Sciences. 363 (1510): 3721–3724. doi:10.1098/rstb.2008.0074. ISSN 0962-8436. PMC 2581657. PMID 18849288.
- Kuru at eMedicine
- Kupfer, L.; Hinrichs, W.; Groschup, M. (1 September 2009). "Prion Protein Misfolding". Current Molecular Medicine. 9 (7): 826–835. doi:10.2174/156652409789105543. PMC 3330701. PMID 19860662.
- Linden, Rafael; Martins, Vilma R.; Prado, Marco A. M.; Cammarota, Martín; Izquierdo, Iván; Brentani, Ricardo R. (April 2008). "Physiology of the Prion Protein". Physiological Reviews. 88 (2): 673–728. doi:10.1152/physrev.00007.2007. PMID 18391177.
- Kuru: The Science and the Sorcery (Siamese Films, 2010)
- Diamond J.M. (1997). Guns, germs, and steel: the fates of human societies. New York: W.W. Norton. p. 208. ISBN 978-0-393-03891-0.
- Liberski, P.P.; Brown, P. (January 2009). "Kuru: Its ramifications after fifty years" (PDF). Experimental Gerontology. 44 (1–2): 63–69. doi:10.1016/j.exger.2008.05.010. PMID 18606515. S2CID 28215397.
- Gibbs, Clarence J.; Amyx, Herbert L.; Bacote, Alfred; Masters, Colin L.; Gajdnsek, D. Carleton (August 1980). "Oral Transmission of Kuru, Creutzfeldt-Jakob Disease, and Scrapie to Nonhuman Primates". The Journal of Infectious Diseases. 142 (2): 205–208. doi:10.1093/infdis/142.2.205. PMID 6997404.
- "Brain disease 'resistance gene' evolves in Papua New Guinea community; could offer insights into CJD". ScienceDaily. 2009-11-21. Retrieved 2016-11-12.
- Mead, Simon; Whitfield, Jerome; Poulter, Mark; Shah, Paresh; Uphill, James; Campbell, Tracy; Al-Dujaily, Huda; Hummerich, Holger; Beck, Jon; Mein, Charles A.; Verzilli, Claudio; Whittaker, John; Alpers, Michael P.; Collinge, John (19 November 2009). "A Novel Protective Prion Protein Variant that Colocalizes with Kuru Exposure" (PDF). New England Journal of Medicine. 361 (21): 2056–2065. doi:10.1056/NEJMoa0809716. PMID 19923577.
- "Natural genetic variation gives complete resistance in prion diseases". Ucl.ac.uk. 2015-06-11. Retrieved 2016-11-12.
- Shirley Lindenbaum (14 Apr 2015). "An annotated history of kuru". Medicine Anthropology Theory.
- "Kuru". Transmissible Spongiform Encephalopathies. Archived from the original on 2016-11-21. Retrieved 2016-11-21.
- Liberski, Paweł P.; Gajos, Agata; Sikorska, Beata; Lindenbaum, Shirley (7 March 2019). "Kuru, the First Human Prion Disease". Viruses. 11 (3): 232. doi:10.3390/v11030232. PMC 6466359. PMID 30866511.
- Liberski, Paweł; Gajos, Agata; Sikorska, Beata; Lindenbaum, Shirley (2019-03-07). "Kuru, the First Human Prion Disease". Viruses. 11 (3): 232. doi:10.3390/v11030232. ISSN 1999-4915. PMC 6466359. PMID 30866511.
- Zigas, Vincent; Gajdusek, Daniel (23 November 1957). "Kuru: Clinical study of a new syndrome resembling paralysis agitans of the eastern highlands of Australian New Guinea". Medical Journal of Australia. 2 (21): 745–754. doi:10.5694/j.1326-5377.1957.tb60287.x. PMID 13492772. S2CID 38647011.
- Kennedy, John (15 May 2012). "Kuru Among the Foré — The Role of Medical Anthropology in Explaining Aetiology and Epidermiology". ArcJohn.wordpress.com. Retrieved 2016-11-21.
- Liberski, P. P.; Brown, P. (2004). "Kuru: A half-opened window onto the landscape of neurodegenerative diseases". Folia Neuropathologica. 42 Suppl A: 3–14. PMID 15449456.
- "Kuru - To Tremble with Fear". Horizon. Season 8. Episode 6. 22 February 1971. BBC2.
- Field, EJ (7 Dec 1967). "The significance of astroglial hypertrophy in Scrapie, Kuru, Multiple Sclerosis and old age together with a note on the possible nature of the scrapie agent". Journal of Neurology. 192 (3): 265–274. doi:10.1007/bf00244170. S2CID 8855940.
- Field, EJ (Feb 1978). "Immunological assessment of ageing: emergence of scrapie-like antigens". Age Ageing. 7 (1): 28–39. doi:10.1093/ageing/7.1.28. PMID 416662.
- Peat, Ray. "BSE - mad cow - scrapie, etc.: Stimulated amyloid degeneration and the toxic fats". RayPeat.com.
Further reading
- Sam Kean. The Tale of the Dueling Neurosurgeons, "Chapter 6: The Laughing Disease", 2014. (Detailed scientific and political history.)