Multiple endocrine neoplasia type 1
Multiple endocrine neoplasia type 1 (MEN-1) is one of a group of disorders, the multiple endocrine neoplasias, that affect the endocrine system through development of neoplastic lesions in pituitary, parathyroid gland and pancreas.[1] It was first described by Paul Wermer in 1954.[2]
| Multiple endocrine neoplasia type 1 | |
|---|---|
| Other names | MEN-1 syndrome |
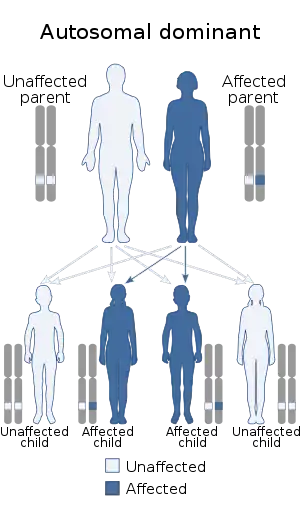 | |
| Multiple endocrine neoplasia type 1 is inherited in an autosomal dominant manner. | |
| Specialty | Oncology |
| Symptoms | Wermer's syndrome |
Signs and symptoms
Parathyroid
Hyperparathyroidism is present in ≥ 90% of patients. Asymptomatic hypercalcemia is the most common manifestation: about 25% of patients have evidence of nephrolithiasis or nephrocalcinosis. In contrast to sporadic cases of hyperparathyroidism, diffuse hyperplasia or multiple adenomas are more common than solitary adenomas.
Pancreas
Pancreatic islet cell tumors are today the major cause of death in persons with MEN-1. Tumors occur in 60-80% of persons with MEN-1 and they are usually multicentric. Multiple adenomas or diffuse islet cell hyperplasia commonly occurs. About 30% of tumors are malignant and have local or distant metastases.[3] About 10-15% of islet cell tumors originate from a β-cell, secrete insulin (insulinoma), and can cause fasting hypoglycemia. β-cell tumors are more common in patients < 40 years of age.
Most islet cell tumors secrete pancreatic polypeptide, the clinical significance of which is unknown. Gastrin is secreted by many non–β-cell tumors (increased gastrin secretion in MEN 1 also often originates from the duodenum). Increased gastrin secretion increases gastric acid, which may inactivate pancreatic lipase, leading to diarrhea and steatorrhea. Increased gastrin secretion also leads to peptic ulcers in > 50% of MEN 1 patients. Usually the ulcers are multiple or atypical in location, and often bleed, perforate, or become obstructed. Peptic ulcer disease may be intractable and complicated. Among patients presenting with Zollinger-Ellison syndrome, 20 to 60% have MEN 1.
A severe secretory diarrhea can develop and cause fluid and electrolyte depletion with non–β-cell tumors. This complex, referred to as the watery diarrhea, hypokalemia and achlorhydria syndrome (VIPoma) has been ascribed to vasoactive intestinal polypeptide, although other intestinal hormones or secretagogues (including prostaglandins) may contribute. Hypersecretion of glucagon, somatostatin, chromogranin, or calcitonin, ectopic secretion of ACTH resulting in Cushing's syndrome, and hypersecretion of somatotropin–releasing hormone (causing acromegaly) sometimes occur in non–β-cell tumors. All of these are rare in MEN 1.Nonfunctioning pancreatic tumors also occur in patients with MEN 1 and may be the most common type of pancreatoduodenal tumor in MEN 1. The size of the nonfunctioning tumor correlates with risk of metastasis and death.
Pituitary
Pituitary tumors occur in 15 to 42% of MEN 1 patients. From 25 to 90% are prolactinomas. About 25% of pituitary tumors secrete growth hormone or growth hormone and prolactin. Excess prolactin may cause galactorrhea, and excess growth hormone causes acromegaly clinically indistinguishable from sporadically occurring acromegaly. About 3% of tumors secrete ACTH, producing Cushing's disease. Most of the remainder are nonfunctional. Local tumor expansion may cause visual disturbance, headache, and hypopituitarism. Pituitary tumors in MEN 1 patients appear to be larger and behave more aggressively than sporadic pituitary tumors.
Other manifestations
Adenomas of adrenal glands occurs occasionally in MEN 1 patients. Hormone secretion is rarely altered as a result, and the significance of these abnormalities is uncertain. Carcinoid tumors, particularly those derived from the embryologic foregut (lungs, thymus), occur in isolated cases. Multiple subcutaneous and visceral lipomas, angiofibromas, and collagenomas may also occur.
Genetic
People with multiple endocrine neoplasia type 1 are born with one mutated copy of the MEN1 gene in each cell. Then, during their lifetime, the other copy of the gene is mutated in a small number of cells. These genetic changes result in no functional copies of the MEN1 gene in selected cells, allowing the cells to divide with little control and form tumors. This is known as Knudson's two-hit hypothesis[4] and is a common feature seen with inherited defects in tumor suppressor genes. Oncogenes can become neoplastic with only one activating mutation, but tumor suppressors inherited from both mother and father must be damaged before they lose their effectiveness. The exception to the "two-hit hypothesis" occurs when suppressor genes exhibit dose-response, such as ATR.[5] The exact function of MEN1 and the protein, menin, produced by this gene is not known,[6] but following the inheritance rules of the "two-hit hypothesis" indicates that it acts as a tumor suppressor.
Diagnosis
In a diagnostic workup individuals with a combination of endocrine neoplasias suggestive of the MEN1 syndrome are recommended to have a mutational analysis of the MEN1 gene if additional diagnostic criteria are sufficiently met, mainly including:[1][7]
- age <40 years
- positive family history
- multifocal or recurrent neoplasia
- two or more organ systems affected
Types
Multiple endocrine neoplasia or MEN is part of a group of disorders that affect the body's network of hormone-producing glands (the endocrine system). Hormones are chemical messengers that travel through the bloodstream and regulate the function of cells and tissues throughout the body. Multiple endocrine neoplasia involves tumors in at least two endocrine glands; tumors can also develop in other organs and tissues. These growths can be noncancerous (benign) or cancerous (malignant). If the tumors become cancerous, some cases can be life-threatening.
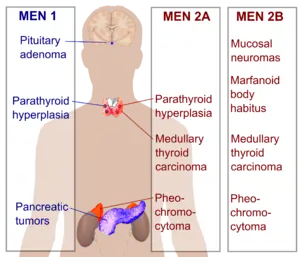
The two major forms of multiple endocrine neoplasia are called type 1 and type 2. These two types are often confused because of their similar names. However, type 1 and type 2 are distinguished by the genes involved,[1] the types of hormones made, and the characteristic signs and symptoms.
These disorders greatly increase the risk of developing multiple cancerous and noncancerous tumors in glands such as the parathyroid, pituitary, and pancreas. Multiple endocrine neoplasia occurs when tumors are found in at least two of the three main endocrine glands (parathyroid, pituitary, and pancreatico-duodenum). Tumors can also develop in organs and tissues other than endocrine glands. If the tumors become cancerous, some cases can be life-threatening. The disorder affects 1 in 30,000 people.
Although many different types of hormone-producing tumors are associated with multiple endocrine neoplasia, tumors of the parathyroid gland, pituitary gland, and pancreas are most frequent in multiple endocrine neoplasia type 1. MEN1-associated overactivity of these three endocrine organs are briefly described here:
- Overactivity of the parathyroid gland (hyperparathyroidism) is the most common sign of this disorder. Hyperparathyroidism disrupts the normal balance of calcium in the blood, which can lead to kidney stones, thinning of the bones (osteoporosis), high blood pressure (hypertension), loss of appetite, nausea, weakness, fatigue, and depression.
- Neoplasia in the pituitary gland can manifest as prolactinomas whereby too much prolactin is secreted, suppressing the release of gonadotropins, causing a decrease in sex hormones such as testosterone. Pituitary tumor in MEN1 can be large and cause signs by compressing adjacent tissues.
- Pancreatic tumors associated with MEN-1 usually form in the beta cells of the islets of Langerhans, causing over-secretion of insulin, resulting in low blood glucose levels (hypoglycemia). However, many other tumors of the pancreatic Islets of Langerhans can occur in MEN-1. One of these, involving the alpha cells, causes over-secretion of glucagon, resulting in a classic triad of high blood glucose levels (hyperglycemia), a rash called necrolytic migratory erythema, and weight loss. Gastrinoma causes the over-secretion of the hormone gastrin, resulting in the over-production of acid by the acid-producing cells of the stomach (parietal cells) and a constellation of sequelae known as Zollinger-Ellison syndrome. Zollinger-Ellison syndrome may include severe gastric ulcers, abdominal pain, loss of appetite, chronic diarrhea, malnutrition, and subsequent weight loss. Other non-beta islet cell tumors associated with MEN1 are discussed below.
Treatment
The treatment of choice of parathyroid tumors is open bilateral exploration with subtotal (3/4) or total parathyroidectomy. Autoimplantation may be considered in case of a total parathyroidectomy. Optimal timing for this operation has not yet been established but it should be performed by an experienced endocrine surgeon.[8]
Endocrine pancreatic tumor are treated with surgery and cytotoxic drugs in case of malignant disease.
Pituitary tumors are treated with surgery (acromegaly and Mb. Cushing) or medicine (prolactinomas).[8]
Culture and society
In the video game Trauma Team, Gabriel Cunningham's son, Joshua Cunningham, is diagnosed with Wermer's syndrome. It is also mentioned in the South Korean drama "Medical Top Team", as Dr. Choi Ah Jin (Oh Yeon-seo) is diagnosed with MEN-1.
See also
References
- Lemmens, I; Van De Ven, W. J.; Kas, K; Zhang, C. X.; Giraud, S; Wautot, V; Buisson, N; De Witte, K; Salandre, J; Lenoir, G; Pugeat, M; Calender, A; Parente, F; Quincey, D; Gaudray, P; De Wit, M. J.; Lips, C. J.; Höppener, J. W.; Khodaei, S; Grant, A. L.; Weber, G; Kytölä, S; Teh, B. T.; Farnebo, F; Thakker, R. V. (1997). "Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1". Human Molecular Genetics. 6 (7): 1177–83. doi:10.1093/hmg/6.7.1177. PMID 9215690.
- WERMER P (1954). "Genetic aspects of adenomatosis of endocrine glands". The American Journal of Medicine. 16 (3): 363–71. doi:10.1016/0002-9343(54)90353-8. PMID 13138607.
- Brandi, Maria Luisa; Agarwal, Sunita K.; Perrier, Nancy D.; Lines, Kate E.; Valk, Gerlof D.; Thakker, Rajesh V. (2021-03-15). "Multiple Endocrine Neoplasia Type 1: Latest Insights". Endocrine Reviews. 42 (2): 133–170. doi:10.1210/endrev/bnaa031. ISSN 1945-7189. PMC 7958143. PMID 33249439.
- Knudson, Alfred G. (1971-04-01). "Mutation and Cancer: Statistical Study of Retinoblastoma". Proceedings of the National Academy of Sciences. 68 (4): 820–823. Bibcode:1971PNAS...68..820K. doi:10.1073/pnas.68.4.820. ISSN 0027-8424. PMC 389051. PMID 5279523.
- Fang, Yanan; Tsao, Cheng-Chung; Goodman, Barbara K; Furumai, Ryohei; Tirado, Carlos A; Abraham, Robert T; Wang, Xiao-Fan (2004-08-04). "ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background". The EMBO Journal. 23 (15): 3164–3174. doi:10.1038/sj.emboj.7600315. ISSN 0261-4189. PMC 514932. PMID 15282542.
- Kumar, Vinay; Abbas, Abul K.; Aster, Jon C. (2014-09-05). Robbins & Cotran Pathologic Basis of Disease. Elsevier Health Sciences. p. 291. ISBN 9780323296359.
- Karges, W.; Schaaf, L.; Dralle, H.; Boehm, B. O. (2000). "Concepts for screening and diagnostic follow-up in multiple endocrine neoplasia type 1 (MEN1)*". Experimental and Clinical Endocrinology & Diabetes. 108 (5): 334–340. doi:10.1055/s-2000-8146. PMID 10989951.
- Thakker R, Newey P, Walls G, Bilezikian J, Dralle H, Ebeling P et al. Clinical Practice Guidelines for Multiple Endocrine Neoplasia Type 1 (MEN1). The Journal of Clinical Endocrinology & Metabolism [Internet]. 2012 [cited 17 January 2020];97(9):2990-3011. Available from: https://doi.org/10.1210/jc.2012-1230
- This article incorporates public domain text from The U.S. National Library of Medicine