Nonsynaptic plasticity
Nonsynaptic plasticity is a form of neuroplasticity that involves modification of ion channel function in the axon, dendrites, and cell body that results in specific changes in the integration of excitatory postsynaptic potentials and inhibitory postsynaptic potentials. Nonsynaptic plasticity is a modification of the intrinsic excitability of the neuron. It interacts with synaptic plasticity, but it is considered a separate entity from synaptic plasticity. Intrinsic modification of the electrical properties of neurons plays a role in many aspects of plasticity from homeostatic plasticity to learning and memory itself. Nonsynaptic plasticity affects synaptic integration, subthreshold propagation, spike generation, and other fundamental mechanisms of neurons at the cellular level. These individual neuronal alterations can result in changes in higher brain function, especially learning and memory. However, as an emerging field in neuroscience, much of the knowledge about nonsynaptic plasticity is uncertain and still requires further investigation to better define its role in brain function and behavior.
Vs. synaptic plasticity
Neuroplasticity is the ability of a particular part or region of a neuron to change in strength over time. There are two largely recognized categories of plasticity: synaptic and nonsynaptic. Synaptic plasticity deals directly with the strength of the connection between two neurons, including amount of neurotransmitter released from the presynaptic neuron, and the response generated in the postsynaptic neuron. Nonsynaptic plasticity involves modification of neuronal excitability in the axon, dendrites, and soma of an individual neuron, remote from the synapse.
Synaptic plasticity
Synaptic plasticity is the ability of a synapse between two neurons to change in strength over time. Synaptic plasticity is caused by changes in use of the synaptic pathway, namely, the frequency of synaptic potentials and the receptors used to relay chemical signals. Synaptic plasticity plays a large role in learning and memory in the brain. Synaptic plasticity can occur through intrinsic mechanisms, in which changes in synapse strength occur because of its own activity, or through extrinsic mechanisms, in which the changes in synapse strength occur via other neural pathways. Short-term inhibitory synaptic plasticity often occurs because of limited neurotransmitter supply at the synapse, and long-term inhibition can occur through decreased receptor expression in the postsynaptic cell. Short-term complementary synaptic plasticity often occurs because of residual or increased ion flow in either the presynaptic or postsynaptic terminal, while long-term synaptic plasticity can occur through the increased production of AMPA and NMDA glutamate receptors, among others, in the postsynaptic cell.[1]
Nonsynaptic plasticity
In comparison, nonsynaptic plasticity is a less well known and somewhat new and ongoing field of research in neuroscience. It is manifested through changes in the characteristics of nonsynaptic structures such as the soma (biology), the axon, or the dendrites. Nonsynaptic plasticity can have short-term or long-term effects. One way these changes occur is through modification of voltage-gated channels in the dendrites and axon, which changes the interpretation of excitatory or inhibitory potentials propagated to the cell. For example, axonal nonsynaptic plasticity can be observed when an action potential fails to reach the presynaptic terminal due to low conduction or buildup of ions. [2]
General excitatory effects
Nonsynaptic and synaptic plasticity have been shown to work concurrently in a variety of ways to produce stimulating effects in the neuron. This includes spike generation, a product of nonsynaptic regulation of potassium and other presynaptic ion channels, which increase the response of the excitatory postsynaptic potential through neurotransmitter release and augmentation of the action potential.[3] Nonsynaptic dendritic plasticity also adds to the effects of synaptic plasticity through widening of the action potential. As will be discussed further, brain-derived neurotrophic factor (BNDF) is produced by neurons to coordinate nonsynaptic and synaptic plasticity.[4] Nonsynaptic changes in the somal body, axon, or dendrites of the neuron are inextricably linked to synaptic strength.
Integration in memory and learning
Although much more is known about the role of synaptic plasticity in memory and learning, both synaptic and nonsynaptic plasticity are essential to memory and learning in the brain. There is much evidence that the two mechanisms both work to achieve the observed effects synergistically. A key example of this is memory formation in the synapse, in which modification of presynaptic release mechanisms and postsynaptic receptors affects either long-term potentiation or depression. Continuous somal depolarization, on the other hand, has been proposed as a method for learned behavior and memory by nonsynaptic plasticity. Nonsynaptic plasticity also augments the effectiveness of synaptic memory formation by regulation of voltage-gated ion channels. Nonsynaptic plasticity is the mechanism responsible for modifications of these channels in the axon, leading to a change in strength of the neuronal action potential, invariably affecting the strength of synaptic mechanisms, and thus the depth and length of memory encoding. [5][6]
Regulation of synaptic plasticity
Nonsynaptic plasticity also has the ability to regulate the effects of synaptic plasticity through negative feedback mechanisms. Change in the number and properties of ion channels in the axon or dendrites has the ability to diminish the effects of a hyperstimulated synapse.[5][6] In the case of extreme overexcitation of these ion channels, backwards flow of ions into the cell will occur, leading to excitotoxicity and cell death by apoptosis or necrosis.[7]
Intrinsic mechanisms
Nonsynaptic neuronal areas such as the axon also have inherent qualities that affect the synapse. These essential mechanisms include the delay in depolarization that action potential undergoes while traveling down the axon. This intrinsic quality slows the propagation of action potentials and is due to the movement of depolarizing current down the cytoplasm and the intermittent placement of sodium channels on the Nodes of Ranvier. These mechanisms always exist, but may change depending on the conditions of the cell soma, axon, and dendrites at the time. Therefore, latency, or delay in propagation of action potentials or excitatory postsynaptic potentials, can be variable. Every excitatory postsynaptic potential that is propagated to a postsynaptic cell is first transmitted through the action potential down the axon in the presynaptic cell, and thus nonsynaptic plasticity inherently affects synaptic plasticity.[1]
Types

Intrinsic excitability of a neuron
The excitability of a neuron at any point depends on the internal and external conditions of the cell at the time of stimulation. Since a neuron typically receives multiple incoming signals at a time, the propagation of an action potential depends on the integration of all the incoming excitatory and inhibitory postsynaptic potentials arriving at the axon hillock. If the summation of all excitatory and inhibitory signals depolarize the cell membrane to the threshold voltage, an action potential is fired. Changing the intrinsic excitability of a neuron will change that neuron's function.
Spike generation
Nonsynaptic plasticity has an excitatory effect on the generation of spikes. The increase in spike generation has been correlated with a decrease in the spike threshold,[3] a response from nonsynaptic plasticity. This response can result from the modulation of certain presynaptic K+ (potassium ion) currents (IA, IK,Ca, and IKs), which work to increase the excitability of the sensory neurons, broaden the action potential, and enhance neurotransmitter release. These modulations of K+ conductances serve as common mechanisms for regulating excitability and synaptic strength.[5]
Regulation of synaptic plasticity
Nonsynaptic plasticity has been linked with synaptic plasticity, via both synergistic and regulatory mechanisms. The degree of synaptic modification determines the polarity of nonsynaptic changes, affecting the change in cellular excitability. Moderate levels of synaptic plasticity produce nonsynaptic changes that will synergistically act with the synaptic mechanisms to strengthen a response. Conversely, more robust levels of synaptic plasticity will produce nonsynaptic responses that will act as a negative feedback mechanism. The negative feedback mechanisms work to protect against saturation or suppression of the circuit activity as a whole.[5]
Axonal modulation
Axonal modulation is a type of plasticity in which the number, activity, or location of ion channels in the axon changes. This causes the neuron to behave differently when stimulated. The modulation of ion channels is a response to a change in the stimulation frequencies of a neuron.
Propagation plasticity
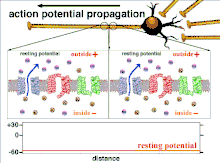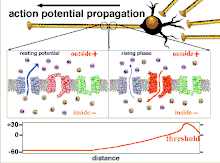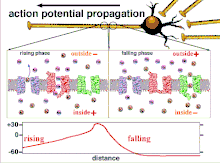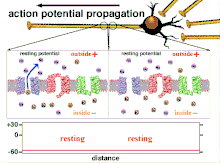
Because it is the summation of the action potentials that eventually results in the threshold polarization being crossed, the temporal relationship of different input signals is very important in determining if and when a post-synaptic neuron will fire. Over time, the time it takes an action potential to propagate down the length of a particular axon can change. In one experiment multielectrode arrays were used to measure the time it took for action potentials to travel from one electrode to another, called latency. The neurons were then stimulated and the value of the latency was recorded over time. The latency values changed over time, suggesting that axonal plasticity influenced the propagation of action potentials.[8]
Shunting
Shunting is a process in which axonal ion channels open during the passive flow (not requiring an ion pump) of a subthreshold depolarization down the axon. Usually occurring at axonal branch points,[9] the timing of these channels opening as the subthreshold signal arrives in the area causes a hyperpolarization to be introduced to the passively flowing depolarization. Therefore, the cell is able to control which branches of the axon the subthreshold depolarization current flows through, resulting in some branches of the axon being more hyperpolarized than others. These differing membrane potentials cause certain areas of the neuron to be more excitable than others, based on the specific location and occurrence of shunting.
High frequency stimulation
Short-term effects: High frequency stimulation of a neuron for a short period of time increases the excitability of the neuron by lowering the amount of voltage required to fire an action potential.[3] High frequency stimulation leads to an increase in the intracellular concentration of sodium and calcium ions due to the repeated opening of voltage-gated sodium and calcium channels in the axon and terminal. As the frequency of stimuli increases, there is less time between each stimulus for the cell to repolarize and return to normal resting potential. Therefore, the resting potential becomes more depolarized, meaning a smaller depolarizing current is needed to fire an action potential.
However, this modulation is usually very short lived. If the stimulation ceases, the neuron will revert to its original resting potential as the ion channels and pumps have ample time to recover from the last stimulus.
Long-term effects: High frequency stimulation of a neuron over a long period of time causes two resulting neuronal changes. Initially, the neuron responds as it would during short-term stimulation, with an increase in excitability. Continuing the high frequency stimulation after this point, results in a drastic, non-reversible change in excitability. When sodium concentrations reach a high enough level in the axon, sodium/calcium pumps reverse their direction of flow, causing calcium to be imported into the cell as sodium is exported out. The increased calcium concentration (and subsequent depolarization of the membrane) inactivates sodium channels and targets them for endocytosis and lysosomal hydrolysis.[10] This results in a major decrease in axonal sodium channels, which are necessary for action potential propagation. If the stimulation continues, eventually the neuron will stop transmitting action potentials and will die. Neuronal death due to overstimulation is called excitotoxicity.
Low frequency stimulation
Short-term effects: All living neurons have a basal rate of action potential propagation and synaptic release. Thus, low frequency stimulation of a neuron in the short term is similar to the activity of a neuron at rest in the brain. No major changes happen to the intrinsic excitability of the neuron.
Long-term effects: Low frequency stimulation of a neuron for a long period of time decreases the excitability of the neuron by activating calcium-dependent phosphatases that tag AMPA receptors for internalization.[11] Low frequency stimulation leads to low levels of calcium in the cell. When calcium concentrations are low, active calcium-dependent phosphatases dominate over calcium-dependent kinases. As more phosphatases are activated, they tag more AMPA receptors for internalization through endocytosis. Since AMPA receptors are one of the main excitatory receptors on neurons, removing them from the cell membrane effectively depresses the cell (if the cell cannot react to excitatory signals, it cannot generate an action potential of its own). In this way low frequency stimulation can actually reverse the effects of long-term potentiation,[12] however these concepts are generally considered types of synaptic plasticity.
Homeostatic and Hebbian plasticity
Central nervous system (CNS) neurons integrate signals from many neurons. In the short term, it is important to have changes in activity of the neuron because this is how information is conveyed in the nervous system (Hebbian plasticity). However, for long-term sustainability, drift towards excitability or inexcitability will disturb the circuit's ability to convey information (homeostatic plasticity). Long-term potentiation (LTP) induces a higher firing rate in post synaptic neurons. It has been hypothesized that the intrinsic properties of a neuron should be arranged to make the most of the dynamic range, acting as a homeostatic mechanism.[13] However, it was shown that intrinsic excitability follows a lognormal distribution which requires active, Hebbian learning to be kept up.[14] In vitro studies have found that when the spontaneous activity of neuronal cultures is inhibited, the neurons become hyper excitable and that when an increase in activity is induced for long periods, the firing rates of the culture drop.[15][16] In contrast, there is a wealth of evidence that the opposite form of regulation, Hebbian learning or LTP-IE/LTD-IE, also occurs[17] and theoretical arguments show that Hebbian plasticity must be the dominant form of plasticity for intrinsic excitability as well.[14] Since homeostatic plasticity also occurs between individual synapses,[18] an earlier view suggesting that homeostatic plasticity and intrinsic plasticity are linked was shown to be inconsistent with evidence.
Mechanism
One mechanism for preserving the dynamic range of a neuron is synaptic scaling, a homeostatic form of plasticity that restores neuronal activity to its normal 'baseline' levels by changing the postsynaptic response of synapses of a neuron as a function of activity. Homeostatic modulation of the intrinsic excitability of a neuron is another way to maintain stability. The regulation of ionic conductances can be achieved in a number of ways, mostly through the release of neuromodulators like dopamine, serotonin etc.[19] Another way is through the controlled release of brain-derived neurotrophic factor (BDNF). BDNF has also been found to influence synaptic scaling, suggesting that this neurotrophic factor may be responsible for the coordination of synaptic and nonsynaptic mechanisms in homeostatic plasticity.[4]
Dendritic excitability
The dendrites are the regions responsible for the integration of the inputs from other neurons. One way that neurons manipulate the integration properties of the dendrites is by changing the number and properties of voltage gated ion channels. Inducing Long-term potentiation (LTP) in a particular synapse, results in an increase in excitability of the dendritic branches specific to that synapse.[20] Dendritic excitability is important for the propagation and integration of synaptic signals. Dendritic excitability is thought to contribute to E-S potentiation, or an increase in the probability that a given input will result in the firing of an action potential.[21]
It is known that changes in dendritic excitability affect action potential back propagation. Action potentials begin near the axon hillock and propagate down the length of the axon, but they also propagate backward through the soma into the dendritic arbor. Active back propagation is dependent on ion channels and changing the densities or properties of these channels can influence the degree to which the signal is attenuated.[21] Plasticity of back-propagation in the dendrites occurs in less than one minute and lasts longer than 25 minutes.[22] Back propagation is a method of signaling to the synapses that an action potential was fired. This is important for spike-timing-dependent plasticity. Fast dendritic adaptation on timescales of few seconds was experimentally observed indicating a potential meaningful global learning mechanism [23][24]
Intrinsic plasticity
Intrinsic plasticity is a form of activity-dependent plasticity distinct from synaptic plasticity, which involves changes at the synapse between two neurons rather than changes in the electrical properties within a single neuron.[25][26] There are some closely related phenomena that can affect a neuron's excitability – such as neuromodulation, structural plasticity, short-term plasticity due to channel kinetics, and neural development.[27][28] There is no consensus on the quantity that intrinsic plasticity regulates, e.g. the firing rate of a neuron, its gain or its internal calcium concentration. Functionally, intrinsic plasticity might allow neurons to learn the intensity of stimuli and represent those intensity statistics in their excitabilities. [29][30] Intrinsic plasticity contributes to encoding memory and complements other forms of activity-dependent plasticity including synaptic plasticity.[31]
Higher brain function
Experimental evidence
The experiment of Kemenes et al.[2] demonstrated that in an extrinsic modulatory neuron, nonsynaptic plasticity influences the expression of long-term associative memory. The relationship between nonsynaptic plasticity and memory was assessed using cerebral giant cells (CGCs). Depolarization from conditioned stimuli increased the neuronal network response. This depolarization lasted as long as the long-term memory. Persistent depolarization and behavioral memory expression occurred more than 24 hours after training, indicating long-term effects. In this experiment, the electrophysiological expression of the long-term memory trace was a conditioned stimulus induced feeding response. CGCs were significantly more depolarized in the trained organisms than the control group, indicating association with learning and excitability changes. When CGCs were depolarized, they showed an increased response to the conditional stimuli and a stronger fictive feeding response. This demonstrated that the depolarization is enough to produce a significant feeding response to the conditioned stimuli. Additionally, no significant difference was observed in the feeding rates between conditioned organisms and ones that were artificially depolarized, reaffirming that depolarization is sufficient to generate the behavior associated with long-term memory.[2]
Memory storage
Nonsynaptic activity in the cell is usually expressed as changes in neuronal excitability. This occurs through modulation of membrane components, such as resting and voltage-gated channels and ion pumps. Nonsynaptic processes are thought to be involved in memory storage. One possible mechanism of this action involves marking a neuron that has been recently active with changes in excitability. This would help to link temporally separated stimuli. Another potential mechanism comes from a computational model that indicates that nonsynaptic plasticity may prime circuits for modification in learning because excitability changes may regulate the threshold for synaptic plasticity.[5]
The storage capacity of synaptic-based memory storage systems is very large, making it an attractive mechanism to study. There are approximately 104 synapses per neuron and 1011 neurons in the human brain.[25] Nonsynaptic plasticity is often overlooked simply because its storage capacity is not as high. Regulating the density of ion channels in the axon and soma of a neuron would change the throughput and affect all of the synapses. Therefore, its storage capacity would be significantly less than that of synaptic plasticity.
While its storage capacity is too low to make it the sole mechanism for storage, nonsynaptic plasticity could contribute to synaptic storage methods. It has been shown that the modulation of ion channels can occur in regions as small as specific dendrites.[20] This specificity makes the storage capacity of nonsynaptic plasticity larger than if it were taken to be whole neuron modulation. Procedural memories are a good fit for this type of storage system because they do not require the high specificity that declarative memories do. Generalization of motor tasks and conditioned stimuli could be an efficient way to store this information.[25]
Learning
Changes in excitability from learning that act as part of the memory trace do so as primers to initiate further changes in the neurons or by a short-term storage mechanism for short-term memory. Nonsynaptic plasticity can emerge during learning as a result of cellular processes, although the timing, persistence, and the relationship between nonsynaptic plasticity and synaptic output are all poorly understood. Studies have shown that nonsynaptic plasticity plays an indirect but important role in the formation of memories. Learning-induced nonsynaptic plasticity is associated with soma depolarization.[5]
Classical conditioning
Experiments have revealed that nonsynaptic changes take place during conditional learning. Woody et al.[32] demonstrated that eyeblink conditioning (EBC), a form of classical conditioning for studying neural structures and mechanisms underlying learning and memory, in a cat is associated with increased excitability and input in the neurons in sensorimotor cortical areas and in the facial nucleus. It was observed that increasing excitability from classical conditioning continued after the response stopped. This suggests that increased excitability may function as a mechanism for memory storage.[5]
In eyeblink conditioning in rabbits, nonsynaptic changes occurred throughout the dorsal hippocampus. This indicates that although excitability changes alone are not enough to explain memory storage processes, nonsynaptic plasticity might be a storage mechanism for phases of memory limited by time. Nonsynaptic changes influence other types of plasticity involved with memory. For example, a nonsynaptic change such as depolarization of the resting membrane potential resulting from conditional learning could cause synaptic plasticity in future learning.[5]
Rule learning and savings
The ability to learn rules is dependent on nonsynaptic plasticity. One study sought to teach rats to discriminate between various odors, and it took several days to teach them to distinguish between a first pair of smells. However, after learning this, the rat was able to learn to distinguish between different odors much faster. Changes in excitability of the pyramidal neurons in these rats were observed for three days after training. These changes faded eventually, suggesting that the neurons were involved in learning the rules, not in storing memory.[5] Daoudal and Debanne attempted to determine if the same learning rules and induction mechanisms defined for synaptic plasticity also applied to nonsynaptic plasticity affecting ion channels. They determined that nonsynaptic and synaptic plasticity share common learning rules and induction pathways, e.g., NMDA receptor dependent long-term potentiation (LTP) and long-term depression (LTD). They also showed that nonsynaptic and synaptic plasticity synergistically form a coherent engram to store memory traces.[22]
Savings is the ability to relearn forgotten information much faster than it was learned originally. Nonsynaptic plasticity is a possible mechanism for this savings effect. During training procedures many neurons experience an increase in intrinsic excitability. This increase in excitability persists even after the memory fades.[5][25]
Substance dependence
Drugs of abuse typically affect the mesolimbic system, or more specifically, the reward pathway of the nervous system. Amongst the common drugs of abuse, nicotine is one of the strongest agonists at the nicotinic cholinergic synapse.[33] Nicotine, competing with acetylcholine (ACh), acts through the nonsynaptic, preterminal, nicotinic acetylcholine receptor (nAChRs) to initiate a membrane potential change and propagate an intracellular Ca2+ signal, thus encouraging the release of neurotransmitters. The specific and characteristic role of calcium current mediated nAChR activity has a different voltage-dependence than other Ca2+ permeable ion channels, as well as different temporal and spatial distribution and as a result, the nonsynaptic nAChR activity enhances the induction of synaptic potentiation, promoting the learning of substance dependence.[34]
Applications to disease
After damage
Nonsynaptic plasticity can function to alleviate the effects of brain damage. When one of the vestibular nerves is damaged, disparity in the firing rates of neurons in the vestibular nuclei causes unnecessary vestibular reflexes. The symptoms of this damage fade over time. This is likely due to modifications of intrinsic excitability in the neurons of the vestibular nucleus.[25][35]
Seizure activity
Nonsynaptic plasticity also plays a key role in seizure activity. Febrile seizures, seizures due to fever early in life, can lead to increased excitability of hippocampal neurons. These neurons become highly sensitized to convulsant agents. It has been shown that seizures early in life can predispose one to more seizures through nonsynaptic mechanisms.[36]
Trauma, including stroke that results in cortical injury, often results in epilepsy. Increased excitability and NMDA conductances result in epileptic activity, suggesting that nonsynaptic plasticity may be the mechanism through which epilepsy is induced after trauma.[37]
Autism
Valproic acid (VPA) is a treatment for epilepsy, migraines, and bipolar disorder that has been linked to many conditions including autism. An animal model of autism exists in which pregnant rats are given VPA. The offspring have traits similar to those of humans with autism. Shortly after birth, these animals exhibit decreased excitability and increased NMDA currents. These effects are corrected at later stages in life. The changes in intrinsic excitability in these animals helped to offset the effects of increased NMDA currents on network activity, a form of homeostatic plasticity. It is believed that this helps mediate the detrimental effects that the increased NMDA currents would have.[38]
Current and future research
Additional research is needed to obtain a broader understanding of nonsynaptic plasticity. Topics that should be further explored as of January 2010 include:
- Local versus global excitability changes in neuronal networks and maintenance of the memory trace[5]
- Specificity of induction of learning-dependent excitability changes[5]
- Manipulation of learning-dependent excitability changes by pharmaceutical products or genetic mutations and their effects on the memory trace[5]
- Similarities between the molecular mechanisms of synaptic and nonsynaptic plasticity[5]
- Comparison of in vivo patterns of nonsynaptic plasticity with in vitro results[5]
- Alterations in gene expression produced by neural activity[39]
References
- Byrne, John H. (1997). "Synaptic Plasticity". Neuroscience Online. The UT Medical School at Houston. Retrieved October 28, 2011.
- Kemenes I, Straub VA, Nikitin ES, Staras K, O'Shea M, Kemenes G, Benjamin PR (July 2006). "Role of delayed nonsynaptic neuronal plasticity in long-term associative memory". Current Biology. 16 (13): 1269–79. doi:10.1016/j.cub.2006.05.049. PMID 16824916. S2CID 16726488.
- Hansel C, Linden DJ, D'Angelo E (May 2001). "Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum". Nature Neuroscience. 4 (5): 467–75. doi:10.1038/87419. PMID 11319554. S2CID 13919286.
- Desai NS, Rutherford LC, Turrigiano GG (1999). "BDNF regulates the intrinsic excitability of cortical neurons". Learning & Memory. 6 (3): 284–91. doi:10.1101/lm.6.3.284. PMC 311300. PMID 10492010.
- Mozzachiodi R, Byrne JH (January 2010). "More than synaptic plasticity: role of nonsynaptic plasticity in learning and memory". Trends in Neurosciences. 33 (1): 17–26. doi:10.1016/j.tins.2009.10.001. PMC 2815214. PMID 19889466.
- Debanne D, Kopysova IL, Bras H, Ferrand N (September 1999). "Gating of action potential propagation by an axonal A-like potassium conductance in the hippocampus: a new type of non-synaptic plasticity". Journal of Physiology, Paris. 93 (4): 285–96. doi:10.1016/S0928-4257(00)80057-1. PMID 10574118. S2CID 30737916.
- Szydlowska K, Tymianski M (February 2010). "Calcium, ischemia and excitotoxicity". Cell Calcium. 47 (2): 122–9. doi:10.1016/j.ceca.2010.01.003. PMID 20167368.
- Bakkum DJ, Chao ZC, Potter SM (May 2008). "Long-term activity-dependent plasticity of action potential propagation delay and amplitude in cortical networks". PLOS ONE. 3 (5): e2088. Bibcode:2008PLoSO...3.2088B. doi:10.1371/journal.pone.0002088. PMC 2324202. PMID 18461127.

- Debanne D, Gähwiler BH, Thompson SM (1996). "Synaptic and non-synaptic plasticity between individual pyramidal cells in the rat hippocampus in vitro". Journal of Physiology, Paris. 90 (5–6): 307–9. doi:10.1016/s0928-4257(97)87903-x. PMID 9089497. S2CID 31639170.
- Ahmed Z, Wieraszko A (January 2009). "Activity-Dependent Axonal Plasticity: The Effects of Electrical Stimulation on Compound Action Potentials Recorded from the Mouse Nervous System In Vitro". The Open Neuroscience Journal. 3 (1): 6. Bibcode:2009ONMJ....1....6P. doi:10.2174/1874082000903010001. S2CID 14646213.
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, et al. (December 2000). "Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization". Nature Neuroscience. 3 (12): 1282–90. doi:10.1038/81814. PMID 11100149. S2CID 18109725.
- O'Dell TJ, Kandel ER (1994). "Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases". Learning & Memory. 1 (2): 129–139. doi:10.1101/lm.1.2.129. PMID 10467591. S2CID 41429873.
- Stemmler M, Koch C (June 1999). "How voltage-dependent conductances can adapt to maximize the information encoded by neuronal firing rate". Nature Neuroscience. 2 (6): 521–7. doi:10.1038/9173. PMID 10448216. S2CID 15134486.
- Scheler G (2017). "Logarithmic distributions prove that intrinsic learning is Hebbian". F1000Research. 6: 1222. arXiv:1410.5610. doi:10.12688/f1000research.12130.2. PMC 5639933. PMID 29071065.
- Corner MA, Ramakers GJ (January 1992). "Spontaneous firing as an epigenetic factor in brain development--physiological consequences of chronic tetrodotoxin and picrotoxin exposure on cultured rat neocortex neurons". Brain Research. Developmental Brain Research. 65 (1): 57–64. doi:10.1016/0165-3806(92)90008-K. PMID 1551233.
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (February 1998). "Activity-dependent scaling of quantal amplitude in neocortical neurons". Nature. 391 (6670): 892–6. Bibcode:1998Natur.391..892T. doi:10.1038/36103. PMID 9495341. S2CID 4328177.
- Mahon S, Charpier S (August 2012). "Bidirectional plasticity of intrinsic excitability controls sensory inputs efficiency in layer 5 barrel cortex neurons in vivo". The Journal of Neuroscience. 32 (33): 11377–89. doi:10.1523/JNEUROSCI.0415-12.2012. PMC 6621180. PMID 22895720.
- Keck T, Keller GB, Jacobsen RI, Eysel UT, Bonhoeffer T, Hübener M (October 2013). "Synaptic scaling and homeostatic plasticity in the mouse visual cortex in vivo". Neuron. 80 (2): 327–34. doi:10.1016/j.neuron.2013.08.018. PMID 24139037. S2CID 13151568.
- Scheler G (2014). "Learning intrinsic excitability in medium spiny neurons". F1000Research. 2: 88. arXiv:q-bio/0502023. doi:10.12688/f1000research.2-88.v2. PMC 4264637. PMID 25520776.
- Frick A, Magee J, Johnston D (February 2004). "LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites". Nature Neuroscience. 7 (2): 126–35. doi:10.1038/nn1178. PMID 14730307. S2CID 11964239.
- Sjöström PJ, Rancz EA, Roth A, Häusser M (April 2008). "Dendritic excitability and synaptic plasticity". Physiological Reviews. 88 (2): 769–840. doi:10.1152/physrev.00016.2007. PMID 18391179. S2CID 1261675.
- Daoudal G, Debanne D (2003). "Long-term plasticity of intrinsic excitability: learning rules and mechanisms". Learning & Memory. 10 (6): 456–65. doi:10.1101/lm.64103. PMID 14657257.
- Hodassman, Shiri; Vardi, Roni; Tugendhaft, Yael; Goldental, Amir; Kanter, Ido (December 2022). "Efficient dendritic learning as an alternative to synaptic plasticity hypothesis". Scientific Reports. 12 (1): 6571. doi:10.1038/s41598-022-10466-8. ISSN 2045-2322. PMC 9051213. PMID 35484180.
- Sardi, Shira; Vardi, Roni; Goldental, Amir; Sheinin, Anton; Uzan, Herut; Kanter, Ido (2018-03-23). "Adaptive nodes enrich nonlinear cooperative learning beyond traditional adaptation by links". Scientific Reports. 8 (1): 5100. Bibcode:2018NatSR...8.5100S. doi:10.1038/s41598-018-23471-7. ISSN 2045-2322. PMC 5865176. PMID 29572466.
- Zhang W, Linden DJ (November 2003). "The other side of the engram: experience-driven changes in neuronal intrinsic excitability". Nature Reviews. Neuroscience. 4 (11): 885–900. doi:10.1038/nrn1248. PMID 14595400. S2CID 17397545.
- Debanne D, Inglebert Y, Russier M (February 2019). "Plasticity of intrinsic neuronal excitability" (PDF). Current Opinion in Neurobiology. 54: 73–82. doi:10.1016/j.conb.2018.09.001. PMID 30243042. S2CID 52812190.
- Triesch, Jochen. "Synergies between intrinsic and synaptic plasticity in individual model neurons." Advances in Neural Information Processing Systems. 2004.
- W. Zhang and D. J. Linden. The other side of the engram: Experience-driven changes in neuronal intrinsic excitability. Nature Reviews Neuroscience, 4:885-900, 2003.
- T. Monk, C. Savin, and J. Lucke. "Optimal neural inference of stimulus intensities." Scientific reports, 8:1, 2018.
- T. Monk, C. Savin, and J. Lucke. Neurons equipped with intrinsic plasticity learn stimulus intensity statistics. Advances in Neural Information Processing Systems. 2016.
- Grasselli G, Boele HJ, Titley HK, Bradford N, van Beers L, Jay L, et al. (January 2020). "SK2 channels in cerebellar Purkinje cells contribute to excitability modulation in motor-learning-specific memory traces". PLOS Biology. 18 (1): e3000596. doi:10.1371/journal.pbio.3000596. PMC 6964916. PMID 31905212.
- Woody CD, Black-Cleworth P (November 1973). "Differences in excitability of cortical neurons as a function of motor projection in conditioned cats". Journal of Neurophysiology. 36 (6): 1104–16. doi:10.1152/jn.1973.36.6.1104. PMID 4761722.
- Kauer JA, Malenka RC (November 2007). "Synaptic plasticity and addiction". Nature Reviews. Neuroscience. 8 (11): 844–58. doi:10.1038/nrn2234. PMID 17948030. S2CID 38811195.
- Dani JA, Ji D, Zhou FM (August 2001). "Synaptic plasticity and nicotine addiction". Neuron. 31 (3): 349–52. doi:10.1016/S0896-6273(01)00379-8. PMID 11516393. S2CID 10062998.
- Darlington CL, Dutia MB, Smith PF (June 2002). "The contribution of the intrinsic excitability of vestibular nucleus neurons to recovery from vestibular damage". The European Journal of Neuroscience. 15 (11): 1719–27. doi:10.1046/j.1460-9568.2002.02024.x. PMID 12081651. S2CID 19939794.
- Bender RA, Dubé C, Gonzalez-Vega R, Mina EW, Baram TZ (2003). "Mossy fiber plasticity and enhanced hippocampal excitability, without hippocampal cell loss or altered neurogenesis, in an animal model of prolonged febrile seizures". Hippocampus. 13 (3): 399–412. doi:10.1002/hipo.10089. PMC 2927853. PMID 12722980.
- Bush PC, Prince DA, Miller KD (October 1999). "Increased pyramidal excitability and NMDA conductance can explain posttraumatic epileptogenesis without disinhibition: a model". Journal of Neurophysiology. 82 (4): 1748–58. doi:10.1152/jn.1999.82.4.1748. PMID 10515964. S2CID 5927906.
- Walcott EC, Higgins EA, Desai NS (September 2011). "Synaptic and intrinsic balancing during postnatal development in rat pups exposed to valproic acid in utero". The Journal of Neuroscience. 31 (37): 13097–109. doi:10.1523/JNEUROSCI.1341-11.2011. PMC 6623264. PMID 21917793.
- Leslie JH, Nedivi E (August 2011). "Activity-regulated genes as mediators of neural circuit plasticity" (PDF). Progress in Neurobiology. 94 (3): 223–37. doi:10.1016/j.pneurobio.2011.05.002. hdl:1721.1/102275. PMC 3134580. PMID 21601615.