PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway is an intracellular signaling pathway important in regulating the cell cycle. Therefore, it is directly related to cellular quiescence, proliferation, cancer, and longevity. PI3K activation phosphorylates and activates AKT, localizing it in the plasma membrane.[1] AKT can have a number of downstream effects such as activating CREB,[2] inhibiting p27,[3] localizing FOXO in the cytoplasm,[3] activating PtdIns-3ps,[4] and activating mTOR[3] which can affect transcription of p70 or 4EBP1.[3] There are many known factors that enhance the PI3K/AKT pathway including EGF,[5] shh,[2] IGF-1,[2] insulin,[3] and CaM.[4] Both leptin and insulin recruit PI3K signalling for metabolic regulation.[6] The pathway is antagonized by various factors including PTEN,[7] GSK3B,[2] and HB9.[5]
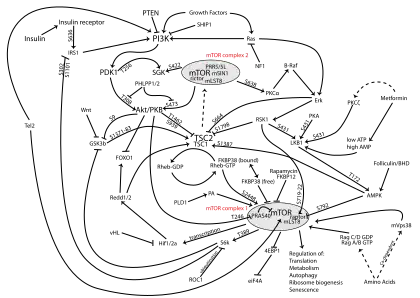
In many cancers, this pathway is overactive, thus reducing apoptosis and allowing proliferation. This pathway is necessary, however, to promote growth and proliferation over differentiation of adult stem cells, neural stem cells specifically.[2] It is the difficulty in finding an appropriate amount of proliferation versus differentiation that researchers are trying to determine in order to utilize this balance in the development of various therapies.[2] Additionally, this pathway has been found to be a necessary component in neural long term potentiation.[4][8]
Proliferation of neural stem cells
Response to glucose
Neural stem cells (NSCs) in the brain must find a balance between maintaining their multipotency by self renewing and proliferating as opposed to differentiating and becoming quiescent. The PI3K/AKT pathway is crucial in this decision making process. NSCs are able to sense and respond to changes in the brain or throughout the organism. When blood glucose levels are elevated acutely, insulin is released from the pancreas. Activation of insulin receptors activates the PI3K/AKT pathway, which promotes proliferation.[3] In this way, when there is high glucose and abundant energy in the organism, the PI3K/AKT pathway is activated and NSCs tend to proliferate. When there are low amounts of available energy, the PI3K/AKT pathway is less active and cells adopt a quiescent state. This occurs, in part, when AKT phosphorylates FOXO, keeping FOXO in the cytoplasm.[3] FOXO, when dephosphorylated, can enter the nucleus and work as a transcription factor to promote the expression of various tumor suppressors such as p27 and p21.[3] These tumor suppressors push the NSC to enter quiescence. FOXO knockouts lose the ability for cells to enter a quiescent state as well as cells losing their neural stem cell character, possibly entering a cancer like state.[3]
PTEN
The PI3K/AKT pathway has a natural inhibitor called Phosphatase and tensin homolog (PTEN) whose function is to limit proliferation in cells, helping to prevent cancer. Knocking out PTEN has been shown to increase the mass of the brain because of the unregulated proliferation that occurs.[3] PTEN works by dephosphorylating PIP3 to PIP2 which limits AKTs ability to bind to the membrane, decreasing its activity. PTEN deficiencies can be compensated downstream to rescue differentiation or quiescence. Knocking out PTEN is not as serious as knocking out FOXO for this reason.[3]
CREB
The cAMP response element CREB is closely related to the cell decision to proliferate or not. Cells that are forced to overexpress AKT increase the amount of CREB and proliferation compared to wild type cells. These cells also express less glial and neural cell markers such as GFAP or β-tubulin.[2] This is because CREB is a transcription factor that influences the transcription of cyclin A which promotes proliferation.[2] For example, adult hippocampal neural progenitor cells need abeyance as stem cells to differentiate later. This is regulated by Shh. Shh works through a slow protein synthesis dependence, which stimulates other cascades that work synergistically with the PI3K/AKT pathway to induce proliferation. Then, the other pathway can be turned off and the effects of the PI3K/AKT pathway become insufficient in stopping differentiation.[2] The specifics of this pathway are unknown.
Roles in cancer
Ovarian cancer
PI3K/ AKT/mTOR pathway is a central regulator of ovarian cancer. PIM kinases are over expressed in many types of cancers and they also contribute to the regulation of ovarian cancer. PIM are directly and indirectly found to activate mTOR and its upstream effectors like AKT. Besides, PIM kinases can cause phosphorylation of IRS, which can alter PI3K. This indicates the close interaction of PIM with PI3K/ AKT/mTOR cascade and its components. Similarly, AKT has also been reported to perform the BAD phosphorylation in OC cells. PIM and the PI3K/AKT/mTOR network both can inhibit the P21 and P27 expressions in OC cells. These data suggest a strong possibility of interaction and relevance of PIM kinases and the PI3K/AKT/mTOR network in the regulation of ovarian cancer.[9] However, targeting this pathway in ovarian cancer has been challenging with several trials failing to achieve sufficient clinical benefit.[10][11]
Breast cancer
In many kinds of breast cancer, aberrations in the PI3K/AKT/mTOR pathway are the most common genomic abnormalities. The most common known aberrations include the PIK3CA gene mutation and the loss-of-function mutations or epigenetic silencing of PTEN.[12] The phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway is activated in approximately 30–40% of BC cases. In triple-negative breast cancer (TNBC), oncogenic activation of the PI3K/AKT/mTOR pathway can happen as a function of overexpression of upstream regulators like EGFR, activating mutations of PIK3CA, loss of function or expression of phosphatase and tensin homolog (PTEN), and the proline-rich inositol polyphosphatase, which are downregulators of PI3K.[13] It is consistent with the hypothesis that PI3K inhibitors can overcome resistance to endocrine therapy when it is acquired
Urothelial cancer
PIK3CA frequently have gain of function mutations in urothelial cancer.[14] Similar to PI3Ka, PI3Kb is expressed in many different cells, and it is mainly involved in the activation of platelets and development of thrombotic diseases. Studies have shown that PI3Kb contribute to tumor proliferation as well. Specifically, it has an important role in tumorigenesis in PTEN-negative cancers.[15] It's reported that interfering with the gene for PI3Kb might be a therapeutic approach for high-risk bladder cancers with mutant PTEN and E-cadherin loss. Specific isoform inhibitors to PI3Kb is a potential treatment for PTEN-deficient cancers.[16]
Prostate cancer
The PI3K pathway is a major source of drug resistance in prostate cancer. This is particularly true in castration-resistant prostate cancer, where tumours become resistant to androgen-deprivation therapy, which block the tumours ability to utilise the hormone androgen to grow.[17] This is due to a complex feedback mechanism which exists between the androgen receptor and the PI3K pathway.[18] As in other tumour types, mutations in key genes of this pathway can lead to hyperactivation of this pathway, for example in PIK3CA,[19][20] Increases in the copy number of PIK3CA and increased mRNA expression also increases pathway activation in prostate cancers among others.[21] Gains in the nearby genetic region 3q26.31-32 have been shown to co-occur with a number of nearby PI3K family members including PIK3CA, PIK3CB and PIK3R4, leading to transcriptional changes in PIK3C2G, PIK3CA, PIK3CB, PIK3R4 as well as pathways associated with cell proliferation.[22] These large spanning gains associate with Gleason grade, tumour stage, lymph node metastasis and other aggressive clinical features.[22] In patients treated with PI3K inhibitors, those with copy number gains in PIK3CB appear to have increased drug susceptibility.[23]
Therapies
PI3K inhibitor
PI3K inhibitors may overcome drug resistance and improve advanced breast cancer (ABC) outcomes.[12] Different PI3K inhibitors exhibit different effect against various PI3K types. Class IA pan-PI3K inhibitors have been more extensively studied than isoform specific inhibitors; Pictilisib is another pan-PI3K inhibitor with greater subunitα-inhibitor activity than buparlisib.[13] Idelalisib is the first PI3K inhibitor approved by the US Food and Drug Administration and is utilized in the treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma and follicular lymphoma. Copanlisib is approved for relapsed follicular lymphoma in patients who have received at least two prior systemic therapies.[24] Duvelisib is approved for relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), and relapsed/refractory follicular lymphoma, both indications for patients who have received at least two prior therapies.[25]
Akt inhibitor
AKT is downstream to PI3K and is inhibited by Ipatasertib.[13] Akt is an AGC-family kinase and a central, integral signaling node of the PAM pathway. There are three Akt isozymes, Akt1, Akt2 and Akt3. Small-molecule inhibitors of Akt1 could be especially useful to target tumors with a high prevalence of Akt1 E17K activating mutations, which is observed in 4–6% of breast cancers and 1–2% of colorectal cancer.[26] Research towards Akt inhibition has focused on inhibition of two distinct binding sites: (1) the allosteric pocket of the inactive enzyme, and (2) the ATP binding site. Allosteric Akt inhibitors, highlighted by MK-2206, have been extensively evaluated in a clinical setting; Recently, additional allosteric Akt inhibitors have been identified. ARQ-092, is a potent pan-Akt inhibitor which can inhibit tumor growth preclinically and is currently in Phase I clinical studies.[26]
mTOR inhibitor
There is significant correlation of phosphorylated mTOR with the survival rate for patients with stages I and II TNBC. A patient-derived xenograft TNBC model testing the mTOR inhibitor rapamycin showed 77–99% tumor-growth inhibition, which is significantly more than has been seen with doxorubicin; protein phosphorylation studies indicated that constitutive activation of the mTOR pathway decreased with treatment.[13]
Dual PI3K/AKT/mTOR inhibitors
It has been hypothesized that blockage of the PI3K/AKT/mTOR pathway can lead to increased antitumor activity in TNBC. Preclinical data have shown that the combination of compounds targeting different cognate molecules in the PI3K/AKT/mTOR pathway leads to synergistic activity. On the basis of these findings, new compounds targeting different components of the PI3K/AKT/mTOR pathway simultaneously continue to be developed. For example, gedatolisib inhibits mutant forms of PI3K-α with elevated kinase activity at concentrations equivalent to the IC50 for wild-type PI3K-α. PI3K-β, -δ and -γ isoforms were inhibited by gedatolisib at concentrations approximately 10-fold higher than those observed for PI3K-α.[13] Another advantage of simultaneously targeting PI3K and mTOR is the ensuing more robust inhibition of receptor tyrosine kinase-positive feedback loops seen with isolated PI3K inhibition.[27] Gedatolisib is currently under development for the treatment of TNBC, in combination with PTK7 antibody–drug conjugate. Apitolisib (GDC-0980) is a PI3K inhibitor (subunits α, δ, and γ) that also targets mTORC [28]
PI3K pathway co-targeted therapy
There are numerous cell signalling pathways that exhibit cross-talk with the PI3K pathway, potentially allowing cancer cells to escape inhibition of PI3K.[29] As such, inhibition of the PI3K pathway alongside other targets could offer a synergistic response, such as that seen with PI3K and MEK co-targeted inhibition in lung cancer cells.[30] More recently, co-targeting the PI3K pathway with PIM kinases has been suggested, with numerous pre-clinical studies suggesting the potential benefit of this approach.[31][32] Development of panels of cell lines that are resistant to inhibition of the PI3K pathway may lead to the identification of future co-targets, and better understanding of which pathways may compensate for loss of PI3K signalling following drug treatment.[33] Combined PI3K inhibition with more traditional therapies such as chemotherapy may also offer improved response over inhibition of PI3K alone.[34]
Neural stem cells
The type of growth factor signaling can effect whether or not NSCs differentiate into motor neurons or not. Priming a media with FGF2 lowers the activity of the PI3K/AKT pathway, which activates GSK3β. This increases expression of HB9.[5] Directly inhibiting PI3K in NSCs leads to a population of cells that are purely HB9+ and differentiate at an elevated efficiency into motor neurons. Grafting these cells into different parts of rats generates motor neurons regardless of the transplanted cells' microenvironment.[5] Following injury, neural stem cells enter a repair phase and express high levels of PI3K to enhance proliferation. This is better for survival of the neurons as a whole but is at the expense of generating motor neurons. Therefore, it can be difficult for injured motor neurons to recover their ability.[5] It is the purpose of modern research to generate neural stem cells that can proliferate but still differentiate into motor neurons. Lowering the effect of the PI3K pathway and increasing the effect of GSK3β and HB9 in NSCs is a potential way of generating these cells.[5]
PTEN inhibitors
PTEN is a tumor suppressor that inhibits the PI3K/AKT pathway. PTEN inhibitors, such as bisperoxovanadium,[35] can enhance the PI3K/AKT pathway to promote cell migration,[36] survival[37] and proliferation.[7] While there are some concerns over possible cell cycle dysregulation and tumorigenesis, temporary and moderate PTEN inhibition may confer neuroprotection against traumatic brain injury[38] and improve CNS recovery by reestablishing lost connections by axonogenesis.[7] Medicinal value of PTEN inhibitors remains to be determined.
Long-term potentiation
In order for long-term potentiation (LTP) to occur, there must be stimulation of NMDA receptors, which causes AMPA receptors to be inserted postsynaptically. PI3K binds to AMPA receptors in a conserved region to orient the receptors in the membrane, specifically at the GluR subunit.[4] PI3K activity increases in response to calcium ions and CaM. Additionally, AKT localizes PtdIns-3Ps in the post synapse, which recruits docking proteins such as tSNARE and Vam7. This directly leads to the docking of AMPA in the post synapse.[4] mTOR activated p70S6K and inactivated 4EBP1 which changes gene expression to allow LTP to occur.[8] Long-term fear conditioning training was affected in rats but there was no effect in short term conditioning. Specifically, amygdala fear conditioning was lost. This is a type of trace conditioning which is a form of learning that requires association of a conditioned stimulus with an unconditioned stimulus. This effect was lost in PI3K knockdowns and increased in PI3K overexpressions.[8]
Role in brain growth
In addition to its role in synaptic plasticity described above, PI3K-AKT signaling pathway also has an important role in brain growth, which is altered when PI3K signaling is disturbed. For example, intracranial volume is also associated with this pathway, in particular with AKT3 intronic variants.[39] Thyroid hormone was originally identified as the primary regulator of brain growth and cognition, and recent evidence has demonstrated that thyroid hormone produces some of its effects on the maturation and plasticity of synapses through PI3K.[40]
See also
- AKT inhibitor
- Akt/PKB signaling pathway
- mTOR inhibitor
- PI3K inhibitor
- PTEN
References
- King D, Yeomanson D, Bryant HE (May 2015). "PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma". Journal of Pediatric Hematology/Oncology. 37 (4): 245–51. doi:10.1097/MPH.0000000000000329. PMID 25811750. S2CID 42323379.
- Peltier J, O'Neill A, Schaffer DV (September 2007). "PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation". Developmental Neurobiology. 67 (10): 1348–61. doi:10.1002/dneu.20506. PMID 17638387. S2CID 16337839.
- Rafalski VA, Brunet A (February 2011). "Energy metabolism in adult neural stem cell fate". Progress in Neurobiology. 93 (2): 182–203. doi:10.1016/j.pneurobio.2010.10.007. PMID 21056618. S2CID 16305263.
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, et al. (May 2003). "Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons". Neuron. 38 (4): 611–24. doi:10.1016/s0896-6273(03)00228-9. PMID 12765612. S2CID 17419450.
- Ojeda L, Gao J, Hooten KG, Wang E, Thonhoff JR, Dunn TJ, et al. (2011). "Critical role of PI3K/Akt/GSK3β in motoneuron specification from human neural stem cells in response to FGF2 and EGF". PLOS ONE. 6 (8): e23414. Bibcode:2011PLoSO...623414O. doi:10.1371/journal.pone.0023414. PMC 3160859. PMID 21887250.
- Garcia-Galiano D, Borges BC, Allen SJ, Elias CF (2019). "PI3K signalling in leptin receptor cells: Role in growth and reproduction". Journal of Neuroendocrinology. 31 (5): e12685. doi:10.1111/jne.12685. PMC 6533139. PMID 30618188.
- Wyatt LA, Filbin MT, Keirstead HS (August 2014). "PTEN inhibition enhances neurite outgrowth in human embryonic stem cell-derived neuronal progenitor cells". The Journal of Comparative Neurology. 522 (12): 2741–55. doi:10.1002/cne.23580. PMID 24610700. S2CID 205683500.
- Sui L, Wang J, Li BM (October 2008). "Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex". Learning & Memory. 15 (10): 762–76. doi:10.1101/lm.1067808. PMID 18832563.
- Aziz AU, Farid S, Qin K, Wang H, Liu B (February 2018). "PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer". Biomolecules. 8 (1): 7. doi:10.3390/biom8010007. PMC 5871976. PMID 29401696.
- Ciccone, Marcia A.; Maoz, Asaf; Casabar, Jennifer K.; Machida, Hiroko; Mabuchi, Seiji; Matsuo, Koji (July 2016). "Clinical outcome of treatment with serine-threonine kinase inhibitors in recurrent epithelial ovarian cancer: a systematic review of literature". Expert Opinion on Investigational Drugs. 25 (7): 781–796. doi:10.1080/13543784.2016.1181748. ISSN 1744-7658. PMC 7534810. PMID 27101098. S2CID 28717797.
- Maoz, Asaf; Ciccone, Marcia A.; Matsuzaki, Shinya; Coleman, Robert L.; Matsuo, Koji (2019-11-22). "Emerging serine-threonine kinase inhibitors for treating ovarian cancer". Expert Opinion on Emerging Drugs. 24 (4): 239–253. doi:10.1080/14728214.2019.1696773. ISSN 1744-7623. PMC 7526049. PMID 31755325. S2CID 208227849.
- Raphael J, Desautels D, Pritchard KI, Petkova E, Shah PS (March 2018). "Phosphoinositide 3-kinase inhibitors in advanced breast cancer: A systematic review and meta-analysis". European Journal of Cancer. 91: 38–46. doi:10.1016/j.ejca.2017.12.010. PMID 29331750.
- Costa RL, Han HS, Gradishar WJ (June 2018). "Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review". Breast Cancer Research and Treatment. 169 (3): 397–406. doi:10.1007/s10549-018-4697-y. PMID 29417298. S2CID 19888056.
- Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. (October 2008). "NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations". Cancer Research. 68 (19): 8022–30. doi:10.1158/0008-5472.CAN-08-1385. PMID 18829560.
- Liu ST, Hui G, Mathis C, Chamie K, Pantuck AJ, Drakaki A (April 2018). "The Current Status and Future Role of the Phosphoinositide 3 Kinase/AKT Signaling Pathway in Urothelial Cancer: An Old Pathway in the New Immunotherapy Era". Clinical Genitourinary Cancer. 16 (2): e269–e276. doi:10.1016/j.clgc.2017.10.011. PMID 29199023. S2CID 4533538.
- Winkler DG, Faia KL, DiNitto JP, Ali JA, White KF, Brophy EE, et al. (November 2013). "PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models". Chemistry & Biology. 20 (11): 1364–74. doi:10.1016/j.chembiol.2013.09.017. PMID 24211136.
- Park, Soonbum; Kim, Young Sik; Kim, Davis Yeon; So, Insuk; Jeon, Ju-Hong (December 2018). "PI3K pathway in prostate cancer: All resistant roads lead to PI3K". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1870 (2): 198–206. doi:10.1016/j.bbcan.2018.09.001. ISSN 1879-2561. PMID 30300679. S2CID 52947672.
- Carver, Brett S.; Chapinski, Caren; Wongvipat, John; Hieronymus, Haley; Chen, Yu; Chandarlapaty, Sarat; Arora, Vivek K.; Le, Carl; Koutcher, Jason; Scher, Howard; Scardino, Peter T. (2011-05-17). "Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer". Cancer Cell. 19 (5): 575–586. doi:10.1016/j.ccr.2011.04.008. ISSN 1878-3686. PMC 3142785. PMID 21575859.
- Zahari, Muhammad Saddiq; Wu, Xinyan; Blair, Brian G.; Pinto, Sneha M.; Nirujogi, Raja S.; Jelinek, Christine A.; Malhotra, Radhika; Kim, Min-Sik; Park, Ben Ho; Pandey, Akhilesh (2015-09-04). "Activating Mutations in PIK3CA Lead to Widespread Modulation of the Tyrosine Phosphoproteome". Journal of Proteome Research. 14 (9): 3882–3891. doi:10.1021/acs.jproteome.5b00302. ISSN 1535-3907. PMC 4641567. PMID 26267517.
- Pearson, Helen B.; Li, Jason; Meniel, Valerie S.; Fennell, Christina M.; Waring, Paul; Montgomery, Karen G.; Rebello, Richard J.; Macpherson, Arthi A.; Koushyar, Sarah; Furic, Luc; Cullinane, Carleen (June 2018). "Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth". Cancer Discovery. 8 (6): 764–779. doi:10.1158/2159-8290.CD-17-0867. ISSN 2159-8290. PMID 29581176.
- Agell, Laia; Hernández, Silvia; Salido, Marta; de Muga, Silvia; Juanpere, Nuria; Arumí-Uria, Montserrat; Menendez, Silvia; Lorenzo, Marta; Lorente, José A.; Serrano, Sergio; Lloreta, Josep (March 2011). "PI3K signaling pathway is activated by PIK3CA mRNA overexpression and copy gain in prostate tumors, but PIK3CA, BRAF, KRAS and AKT1 mutations are infrequent events". Modern Pathology. 24 (3): 443–452. doi:10.1038/modpathol.2010.208. ISSN 1530-0285. PMID 21113138. S2CID 27405431.
- Simpson, Benjamin S.; Camacho, Niedzica; Luxton, Hayley J.; Pye, Hayley; Finn, Ron; Heavey, Susan; Pitt, Jason; Moore, Caroline M.; Whitaker, Hayley C. (2020-08-14). "Genetic alterations in the 3q26.31-32 locus confer an aggressive prostate cancer phenotype". Communications Biology. 3 (1): 440. doi:10.1038/s42003-020-01175-x. ISSN 2399-3642. PMC 7429505. PMID 32796921. S2CID 221118233.
- Bono, Johann de; Arkenau, Hendrik-Tobias; Mateo, Joaquin; Infante, Jeffrey R.; Burris, Howard A.; Bang, Yung-Jue; Eder, Joseph; Sharma, Sunil; Chung, Hyun C.; Decordova, Shaun; Swales, Karen E. (2015-08-01). "Abstract CT328: Exploratory genetic analysis of tumors from a phase I/II dose escalation study of GSK2636771 in patients (pts) with PTEN deficient advanced tumors". Cancer Research. 75 (15 Supplement): CT328. doi:10.1158/1538-7445.AM2015-CT328. ISSN 0008-5472.
- Greenwell IB, Ip A, Cohen JB (November 2017). "PI3K Inhibitors: Understanding Toxicity Mechanisms and Management". Oncology. 31 (11): 821–8. PMID 29179250.
- "duvelisib (COPIKTRA, Verastem, Inc.) for adult patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL)". U.S. Food and Drug Administration. Retrieved 23 October 2018.
- Huck BR, Mochalkin I (July 2017). "Recent progress towards clinically relevant ATP-competitive Akt inhibitors". Bioorganic & Medicinal Chemistry Letters. 27 (13): 2838–2848. doi:10.1016/j.bmcl.2017.04.090. PMID 28506751.
- Chakrabarty A, Sánchez V, Kuba MG, Rinehart C, Arteaga CL (February 2012). "Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors". Proceedings of the National Academy of Sciences of the United States of America. 109 (8): 2718–23. Bibcode:2012PNAS..109.2718C. doi:10.1073/pnas.1018001108. PMC 3286932. PMID 21368164.
- Cappellen D, Gil Diez de Medina S, Chopin D, Thiery JP, Radvanyi F (June 1997). "Frequent loss of heterozygosity on chromosome 10q in muscle-invasive transitional cell carcinomas of the bladder". Oncogene. 14 (25): 3059–66. doi:10.1038/sj.onc.1201154. PMID 9223669.
- Heavey, Susan; O'Byrne, Kenneth J.; Gately, Kathy (April 2014). "Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC". Cancer Treatment Reviews. 40 (3): 445–456. doi:10.1016/j.ctrv.2013.08.006. ISSN 1532-1967. PMID 24055012.
- Heavey, Susan; Cuffe, Sinead; Finn, Stephen; Young, Vincent; Ryan, Ronan; Nicholson, Siobhan; Leonard, Niamh; McVeigh, Niall; Barr, Martin; O'Byrne, Kenneth; Gately, Kathy (2016-11-29). "In pursuit of synergy: An investigation of the PI3K/mTOR/MEK co-targeted inhibition strategy in NSCLC". Oncotarget. 7 (48): 79526–79543. doi:10.18632/oncotarget.12755. ISSN 1949-2553. PMC 5346733. PMID 27765909.
- Luszczak, Sabina; Kumar, Christopher; Sathyadevan, Vignesh Krishna; Simpson, Benjamin S.; Gately, Kathy A.; Whitaker, Hayley C.; Heavey, Susan (2020). "PIM kinase inhibition: co-targeted therapeutic approaches in prostate cancer". Signal Transduction and Targeted Therapy. 5: 7. doi:10.1038/s41392-020-0109-y. ISSN 2059-3635. PMC 6992635. PMID 32025342.
- Malone, Tom; Schäfer, Lea; Simon, Nathalie; Heavey, Susan; Cuffe, Sinead; Finn, Stephen; Moore, Gillian; Gately, Kathy (March 2020). "Current perspectives on targeting PIM kinases to overcome mechanisms of drug resistance and immune evasion in cancer" (PDF). Pharmacology & Therapeutics. 207: 107454. doi:10.1016/j.pharmthera.2019.107454. ISSN 1879-016X. PMID 31836451. S2CID 209357486.
- Heavey, Susan; Dowling, Paul; Moore, Gillian; Barr, Martin P.; Kelly, Niamh; Maher, Stephen G.; Cuffe, Sinead; Finn, Stephen P.; O'Byrne, Kenneth J.; Gately, Kathy (26 January 2018). "Development and characterisation of a panel of phosphatidylinositide 3-kinase - mammalian target of rapamycin inhibitor resistant lung cancer cell lines". Scientific Reports. 8 (1): 1652. Bibcode:2018NatSR...8.1652H. doi:10.1038/s41598-018-19688-1. ISSN 2045-2322. PMC 5786033. PMID 29374181.
- Heavey, Susan; Godwin, Peter; Baird, Anne-Marie; Barr, Martin P.; Umezawa, Kazuo; Cuffe, Sinéad; Finn, Stephen P.; O'Byrne, Kenneth J.; Gately, Kathy (October 2014). "Strategic targeting of the PI3K-NFκB axis in cisplatin-resistant NSCLC". Cancer Biology & Therapy. 15 (10): 1367–1377. doi:10.4161/cbt.29841. ISSN 1555-8576. PMC 4130730. PMID 25025901.
- Schmid AC, Byrne RD, Vilar R, Woscholski R (May 2004). "Bisperoxovanadium compounds are potent PTEN inhibitors". FEBS Letters. 566 (1–3): 35–8. doi:10.1016/j.febslet.2004.03.102. PMID 15147864.
- Mihai C, Bao S, Lai JP, Ghadiali SN, Knoell DL (February 2012). "PTEN inhibition improves wound healing in lung epithelia through changes in cellular mechanics that enhance migration". American Journal of Physiology. Lung Cellular and Molecular Physiology. 302 (3): L287-99. doi:10.1152/ajplung.00037.2011. PMC 3289272. PMID 22037358.
- Lai JP, Dalton JT, Knoell DL (December 2007). "Phosphatase and tensin homologue deleted on chromosome ten (PTEN) as a molecular target in lung epithelial wound repair". British Journal of Pharmacology. 152 (8): 1172–84. doi:10.1038/sj.bjp.0707501. PMC 2189995. PMID 17922022.
- Walker CL, Walker MJ, Liu NK, Risberg EC, Gao X, Chen J, Xu XM (2012). "Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury". PLOS ONE. 7 (1): e30012. Bibcode:2012PLoSO...730012W. doi:10.1371/journal.pone.0030012. PMC 3254642. PMID 22253859.
- Adams HH, Hibar DP, Chouraki V, Stein JL, Nyquist PA, Rentería ME, et al. (December 2016). "Novel genetic loci underlying human intracranial volume identified through genome-wide association". Nature Neuroscience. 19 (12): 1569–1582. doi:10.1038/nn.4398. PMC 5227112. PMID 27694991.
- Martin NP, Marron Fernandez de Velasco E, Mizuno F, Scappini EL, Gloss B, Erxleben C, et al. (September 2014). "A rapid cytoplasmic mechanism for PI3 kinase regulation by the nuclear thyroid hormone receptor, TRβ, and genetic evidence for its role in the maturation of mouse hippocampal synapses in vivo". Endocrinology. 155 (9): 3713–24. doi:10.1210/en.2013-2058. PMC 4138568. PMID 24932806.