Parathyroid hormone
Parathyroid hormone (PTH), also called parathormone or parathyrin, is a peptide hormone secreted by the parathyroid glands that regulates the serum calcium concentration through its effects on bone, kidney, and intestine.[5]





PTH influences bone remodeling, which is an ongoing process in which bone tissue is alternately resorbed and rebuilt over time. PTH is secreted in response to low blood serum calcium (Ca2+) levels. PTH indirectly stimulates osteoclast activity within the bone matrix (osteon), in an effort to release more ionic calcium (Ca2+) into the blood to elevate a low serum calcium level. The bones act as a (metaphorical) "bank of calcium" from which the body can make "withdrawals" as needed to keep the amount of calcium in the blood at appropriate levels despite the ever-present challenges of metabolism, stress, and nutritional variations. PTH is "a key that unlocks the bank vault" to remove the calcium.
PTH is secreted primarily by the chief cells of the parathyroid glands. The gene for PTH is located on chromosome 11. It is a polypeptide containing 84 amino acids, which is a prohormone. It has a molecular mass around 9500 Da.[6] Its action is opposed by the hormone calcitonin.
There are two types of PTH receptors. Parathyroid hormone 1 receptors, activated by the 34 N-terminal amino acids of PTH, are present at high levels on the cells of bone and kidney. Parathyroid hormone 2 receptors are present at high levels on the cells of central nervous system, pancreas, testes, and placenta.[7] The half-life of PTH is about 4 minutes.[8]
Disorders that yield too little or too much PTH, such as hypoparathyroidism, hyperparathyroidism, and paraneoplastic syndromes can cause bone disease, hypocalcemia, and hypercalcemia.
Structure
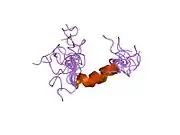
hPTH-(1-84) crystallizes as a slightly bent, long, helical dimer. The extended helical conformation of hPTH-(1-84) is the likely bioactive conformation.[9] The N-terminal fragment 1-34 of parathyroid hormone (PTH) has been crystallized and the structure has been refined to 0.9 Å resolution.
 helical dimer structure of hPTH-(1-34)[10] |
Function
Regulation of serum calcium

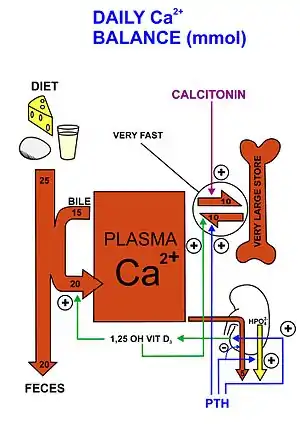
Parathyroid hormone regulates serum calcium through its effects on bone, kidney, and the intestine:[5]
In bone, PTH enhances the release of calcium from the large reservoir contained in the bones.[16] Bone resorption is the normal destruction of bone by osteoclasts, which are indirectly stimulated by PTH. Stimulation is indirect since osteoclasts do not have a receptor for PTH; rather, PTH binds to osteoblasts, the cells responsible for creating bone. Binding stimulates osteoblasts to increase their expression of RANKL and inhibits their secretion of osteoprotegerin (OPG). Free OPG competitively binds to RANKL as a decoy receptor, preventing RANKL from interacting with RANK, a receptor for RANKL. The binding of RANKL to RANK (facilitated by the decreased amount of OPG available for binding the excess RANKL) stimulates osteoclast precursors, which are of a monocyte lineage, to fuse. The resulting multinucleated cells are osteoclasts, which ultimately mediate bone resorption. Estrogen also regulates this pathway through its effects on PTH. Estrogen suppresses T cell TNF production by regulating T cell differentiation and activity in the bone marrow, thymus, and peripheral lymphoid organs. In the bone marrow, estrogen downregulates the proliferation of hematopoietic stem cells through an IL-7 dependent mechanism.[17]
In the kidney, around 250 mmol of calcium ions are filtered into the glomerular filtrate per day. Most of this (245 mmol/d) is reabsorbed from the tubular fluid, leaving about 5 mmol/d to be excreted in the urine. This reabsorption occurs throughout the tubule (most, 60-70%, of it in the proximal tubule), except in the thin segment of the loop of Henle.[11] Circulating parathyroid hormone only influences the reabsorption that occurs in the distal tubules and the renal collecting ducts[11] (but see Footnote[nb 1]). A more important effect of PTH on the kidney is, however, its inhibition of the reabsorption of phosphate (HPO42−) from the tubular fluid, resulting in a decrease in the plasma phosphate concentration. Phosphate ions form water-insoluble salts with calcium. Thus, a decrease in the phosphate concentration of the blood plasma (for a given total calcium concentration) increases the amount of calcium that is ionized.[20][21] A third important effect of PTH on the kidney is its stimulation of the conversion of 25-hydroxy vitamin D into 1,25-dihydroxy vitamin D (calcitriol), which is released into the circulation. This latter form of vitamin D is the active hormone which stimulates calcium uptake from the intestine.[22]
Via the kidney, PTH enhances the absorption of calcium in the intestine by increasing the production of activated vitamin D. Vitamin D activation occurs in the kidney. PTH up-regulates 25-hydroxyvitamin D3 1-alpha-hydroxylase, the enzyme responsible for 1-alpha hydroxylation of 25-hydroxy vitamin D, converting vitamin D to its active form (1,25-dihydroxy vitamin D). This activated form of vitamin D increases the absorption of calcium (as Ca2+ ions) by the intestine via calbindin.
PTH was one of the first hormones to be shown to use the G-protein adenylyl cyclase second messenger system.
Regulation of serum phosphate
PTH reduces the reabsorption of phosphate from the proximal tubule of the kidney,[23] which means more phosphate is excreted through the urine.
However, PTH enhances the uptake of phosphate from the intestine and bones into the blood. In the bone, slightly more calcium than phosphate is released from the breakdown of bone. In the intestines, absorption of both calcium and phosphate is mediated by an increase in activated vitamin D. The absorption of phosphate is not as dependent on vitamin D as is that of calcium. The end result of PTH release is a small net drop in the serum concentration of phosphate.
Vitamin D synthesis
PTH upregulates the activity of 1-α-hydroxylase enzyme, which converts 25-hydroxycholecalciferol, the major circulating form of inactive vitamin D, into 1,25-dihydroxycholecalciferol, the active form of vitamin D, in the kidney.
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]

- The interactive pathway map can be edited at WikiPathways: "VitaminDSynthesis_WP1531".
Regulation of PTH secretion
Secretion of parathyroid hormone is determined chiefly by serum ionized calcium concentration through negative feedback. Parathyroid cells express calcium-sensing receptors on the cell surface. PTH is secreted when [Ca2+] is decreased (calcitonin is secreted when serum calcium levels are elevated). The G-protein-coupled calcium receptors bind extracellular calcium and may be found on the surface on a wide variety of cells distributed in the brain, heart, skin, stomach, C cells, and other tissues. In the parathyroid gland, high concentrations of extracellular calcium result in activation of the Gq G-protein coupled cascade through the action of phospholipase C. This hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to liberate intracellular messengers IP3 and diacylglycerol (DAG). Ultimately, these two messengers result in a release of calcium from intracellular stores into the cytoplasmic space. Hence a high extracellular calcium concentration leads to an increase in the cytoplasmic calcium concentration. In contrast to the mechanism that most secretory cells use, this high cytoplasmic calcium concentration inhibits the fusion of vesicles containing granules of preformed PTH with the membrane of the parathyroid cell, and thus inhibits release of PTH.
In the parathyroids, magnesium serves this role in stimulus-secretion coupling. A mild decrease in serum magnesium levels stimulates the reabsorptive activity PTH has on the kidneys. Severe hypomagnesemia inhibits PTH secretion and also causes resistance to PTH, leading to a form of hypoparathyroidism that is reversible.[24]
Stimulators
- Decreased serum [Ca2+].
- Mild decreases in serum [Mg2+].
- An increase in serum phosphate (increased phosphate causes it to complex with serum calcium, forming calcium phosphate, which reduces stimulation of Ca-sensitive receptors (CaSr) that do not sense calcium phosphate, triggering an increase in PTH).
- Adrenaline
- Histamine
Inhibitors
- Increased serum [Ca2+].
- Severe decreases in serum [Mg2+], which also produces symptoms of hypoparathyroidism (such as hypocalcemia).[25]
- Calcitriol
- Increase in serum phosphate. Fibroblast growth factor-23 (FGF23) is produced in osteoblasts (from bone) in response to increases in serum phosphate (Pi). It binds to the fibroblast growth factor receptor of the parathyroid and suppresses PTH release. This may seem contradictory because PTH actually helps rid the blood of phosphates but it is also causes release of phosphate into the blood from bone resorption. FGF23 inhibits PTH and then takes its place helping inhibit re-absorption of phosphate in the kidney without the phosphate releasing effect on bones.[26][27]
Disorders
Hyperparathyroidism, the presence of excessive amounts of parathyroid hormone in the blood, occurs in two very distinct sets of circumstances. Primary hyperparathyroidism is due to autonomous, abnormal hypersecretion of PTH from the parathyroid gland, while secondary hyperparathyroidism is an appropriately high PTH level seen as a physiological response to hypocalcemia. A low level of PTH in the blood is known as hypoparathyroidism and is most commonly due to damage to or removal of parathyroid glands during thyroid surgery.
There are a number of rare but well-described genetic conditions affecting parathyroid hormone metabolism, including pseudohypoparathyroidism, familial hypocalciuric hypercalcemia, and autosomal dominant hypercalciuric hypocalcemia. Of note, PTH is unchanged in pseudopseudohypoparathyroidism. In osteoporotic women, administration of an exogenous parathyroid hormone analogue (teriparatide, by daily injection) superimposed on estrogen therapy produced increases in bone mass and reduced vertebral and nonvertebral fractures by 45 to 65%.[28]
Measurement
PTH can be measured in the blood in several different forms: intact PTH; N-terminal PTH; mid-molecule PTH, and C-terminal PTH, and different tests are used in different clinical situations. The level may be stated in pg/dL or pmol/L (sometimes abbreviated mmol/L); multiply by 0.1060 to convert from pg/dL to pmol/L.[29]
A US source states the average PTH level to be 8–51 pg/mL.[30] In the UK the biological reference range is considered to be 1.6-6.9 pmol/L.[31] Normal total plasma calcium level ranges from 8.5 to 10.2 mg/dL (2.12 mmol/L to 2.55 mmol/L).[32]
Interpretive guide
The intact PTH and calcium normal ranges are different for age; calcium is also different for sex.[33][34]
| Condition | Intact PTH | Calcium |
|---|---|---|
| Normal Parathyroid | Normal | Normal |
| Hypoparathyroidism | Low or Low Normal [note 1] | Low |
| Hyperparathyroidism | ||
| - Primary | High or Normal [note 1] | High |
| - Secondary | High | Normal or Low |
| - Tertiary[note 2] | High | High |
| Non-Parathyroid Hypercalcemia | Low or Low Normal [note 1] | High |
- Low Normal or Normal only for Quest Lab, not LabCorp
- Both primary and tertiary hyperparathyroidism may have high PTH and high calcium. Tertiary is differentiated from primary hyperparathyroidism by a history of chronic kidney failure and secondary hyperparathyroidism.
Model organisms
Model organisms have been used in the study of PTH function. A conditional knockout mouse line called Pthtm1a(EUCOMM)Wtsi was generated at the Wellcome Trust Sanger Institute.[35] Male and female animals underwent a standardized phenotypic screen[36] to determine the effects of deletion.[37][38][39][40] Additional screens performed: - In-depth immunological phenotyping[41]
| Characteristic | Phenotype |
|---|---|
| All data available at.[36][41] | |
| Insulin | Normal |
| Homozygous viability at P14 | Normal |
| Homozygous Fertility | Normal |
| Body weight | Normal |
| Neurological assessment | Normal |
| Grip strength | Normal |
| Dysmorphology | Normal |
| Indirect calorimetry | Normal |
| Glucose tolerance test | Normal |
| Auditory brainstem response | Normal |
| DEXA | Normal |
| Radiography | Normal |
| Eye morphology | Normal |
| Clinical chemistry | Abnormal |
| Haematology 16 Weeks | Normal |
| Peripheral blood leukocytes 16 Weeks | Normal |
| Salmonella infection | Normal |
See also
Footnote
- This reduction in the rate of calcium excretion via the urine is a minor effect of high parathyroid hormone levels in the blood. The main determinant of the amount of calcium excreted into the urine per day is the plasma ionized calcium concentration itself. The plasma parathyroid hormone (PTH) concentration only increases or decreases the amount of calcium excreted at any specified plasma ionized calcium concentration. Thus, in primary hyperparathyroidism, the quantity of calcium excreted in the urine per day is increased despite the high levels of PTH in the blood, because hyperparathyroidism results in hypercalcemia, which increases the urinary calcium concentration (hypercalcuria) despite the moderately increased rate of calcium reabsorption from the renal tubular fluid caused by PTH's direct effect on those tubules. Renal stones are, therefore, often a first indication of hyperparathyroidism, especially since the hypercalcuria is accompanied by an increase in urinary phosphate excretion (a direct result of the high plasma PTH levels). Together the calcium and phosphate tend to precipitate out as water-insoluble salts, which readily form solid “stones”.[11][18][19]
References
- GRCh38: Ensembl release 89: ENSG00000152266 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000059077 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Coetzee M, Kruger MC (May 2004). "Osteoprotegerin-receptor activator of nuclear factor-kappaB ligand ratio: a new approach to osteoporosis treatment?". Southern Medical Journal. 97 (5): 506–11. doi:10.1097/00007611-200405000-00018. PMID 15180028. S2CID 45131847.
- Brewer HB, Fairwell T, Ronan R, Sizemore GW, Arnaud CD (1972). "Human parathyroid hormone: amino-acid sequence of the amino-terminal residues 1-34". Proceedings of the National Academy of Sciences of the United States of America. 69 (12): 3585–8. Bibcode:1972PNAS...69.3585B. doi:10.1073/pnas.69.12.3585. PMC 389826. PMID 4509319.
- Nosek, Thomas M. "Section 5/5ch6/s5ch6_11". Essentials of Human Physiology. Archived from the original on 2016-03-24.
- Bieglmayer C, Prager G, Niederle B (Oct 2002). "Kinetic analyses of parathyroid hormone clearance as measured by three rapid immunoassays during parathyroidectomy". Clinical Chemistry. 48 (10): 1731–8. doi:10.1093/clinchem/48.10.1731. PMID 12324490. Archived from the original on 2011-06-07. Retrieved 2009-02-23.
- Jin L, Briggs SL, Chandrasekhar S, Chirgadze NY, Clawson DK, Schevitz RW, Smiley DL, Tashjian AH, Zhang F (Sep 2000). "Crystal structure of human parathyroid hormone 1-34 at 0.9-A resolution". The Journal of Biological Chemistry. 275 (35): 27238–44. doi:10.1074/jbc.M001134200. PMID 10837469.
- PDB: 1ETE; Savvides SN, Boone T, Andrew Karplus P (Jun 2000). "Flt3 ligand structure and unexpected commonalities of helical bundles and cystine knots". Nature Structural Biology. 7 (6): 486–91. doi:10.1038/75896. PMID 10881197.
- Blaine J, Chonchol M, Levi M (2015). "Renal control of calcium, phosphate, and magnesium homeostasis". Clinical Journal of the American Society of Nephrology. 10 (7): 1257–72. doi:10.2215/CJN.09750913. PMC 4491294. PMID 25287933.
- Brini M, Ottolini D, Calì T, Carafoli E (2013). "Chapter 4. Calcium in Health and Disease". In Sigel A, Helmut RK (eds.). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. Vol. 13. Springer. pp. 81–137. doi:10.1007/978-94-007-7500-8_4. PMID 24470090.
- Walter F (2003). "The Parathyroid Glands and Vitamin D". Medical Physiology: A Cellular And Molecular Approach. Elsevier/Saunders. p. 1094. ISBN 1-4160-2328-3.
- Guyton A (1976). ‘’Medical Physiology’’. p.1062; New York, Saunders and Co.
- Barrett KE, Barman SM, Boitano S, Brooks H. "Chapter 23. Hormonal Control of Calcium & Phosphate Metabolism & the Physiology of Bone". In Barrett KE, Barman SM, Boitano S, Brooks H (eds.). Ganong's Review of Medical Physiology (23e ed.). Archived from the original on 2011-07-07. Retrieved 2016-01-03.
- Poole KE, Reeve J (Dec 2005). "Parathyroid hormone - a bone anabolic and catabolic agent". Current Opinion in Pharmacology. 5 (6): 612–7. doi:10.1016/j.coph.2005.07.004. PMID 16181808.
- Bord S, Ireland DC, Beavan SR, Compston JE (2003). "The effects of estrogen on osteoprotegerin, RANKL, and estrogen receptor expression in human osteoblasts". Bone. 32 (2): 136–41. doi:10.1016/S8756-3282(02)00953-5. PMID 12633785.
- Harrison TR, Adams RD, Bennett IL, Resnick WH, Thorn GW, Wintrobe MM (1958). "Metabolic and Endocrine Disorders". Principles of Internal Medicine (Third ed.). New York: McGraw-Hill Book Company. pp. 575–578.
- "Symptoms of Hyperparathyroidism and Symptoms of Parathyroid Disease". Parathyroid.com. Norman Parathyroid Center. Retrieved 2015-12-30.
- Haldimann B, Vogt K (1983). "[Hyperphosphatemia and tetany following phosphate enema]". Schweizerische Medizinische Wochenschrift (in French). 113 (35): 1231–3. PMID 6623048.
- Sutters M, Gaboury CL, Bennett WM (1996). "Severe hyperphosphatemia and hypocalcemia: a dilemma in patient management". Journal of the American Society of Nephrology. 7 (10): 2056–61. doi:10.1681/ASN.V7102056. PMID 8915965.
- Stryer L (1995). Biochemistry (Fourth ed.). New York: W.H. Freeman and Company. p. 707. ISBN 978-0-7167-2009-6.
- Gardner D, Shoback D (2011). Greenspan's Basic & Clinical Endocrinology (9th ed.). McGraw Hill. p. 232. ISBN 978-0-07-162243-1.
- Agus ZS (Jul 1999). "Hypomagnesemia". Journal of the American Society of Nephrology. 10 (7): 1616–22. doi:10.1681/ASN.V1071616. PMID 10405219. Archived from the original on 2016-12-10. Retrieved 2013-08-12.
- Costanzo, Linda S. (2007). BRS Physiology. Lippincott, Williams, & Wilkins. pp. 260. ISBN 978-0-7817-7311-9.
- Blaine J, Chonchol M, Levi M (July 2015). "Renal control of calcium, phosphate, and magnesium homeostasis". Clinical Journal of the American Society of Nephrology. 10 (7): 1257–72. doi:10.2215/CJN.09750913. PMC 4491294. PMID 25287933.
- Carrillo-López N, Fernández-Martín JL, Cannata-Andía JB (2009-04-01). "[The role of calcium, calcitriol and their receptors in parathyroid regulation]". Nefrologia. 29 (2): 103–8. doi:10.3265/Nefrologia.2009.29.2.5154.en.full. PMID 19396314.
- Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH (May 2001). "Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis". The New England Journal of Medicine. 344 (19): 1434–41. doi:10.1056/NEJM200105103441904. PMID 11346808.
- "Parathyroid hormone (PTH) unit conversion (online calculator)". Unitslab.
- Longo DL, Fauci A, Kasper D, Hauser S, Jameson J, Loscalzo J (2012). Harrison's Principles of Internal Medicine (18th ed.). New York: McGraw-Hill. p. 3594. ISBN 978-0-07-174889-6.
- "Division of Laboratory Medicine: Parathyroid hormone" (PDF). Manchester University NHS Foundation Trust (UK).
- Zieve D. "MedlinePlus Medical Encyclopedia: Serum calcium". National Library of Medicine, National Institutes of Health. Retrieved 2009-02-01.
- PTH, Intact and Calcium Test Detail. Quest Diagnostics Lab. Accessed 2019-06-29.
- Parathyroid Hormone (PTH) Plus Calcium. LabCorp. Accessed 2019-07-02.
- Gerdin AK (2010). "The Sanger Mouse Genetics Programme: high throughput characterisation of knockout mice". Acta Ophthalmologica. 88: 925–7. doi:10.1111/j.1755-3768.2010.4142.x. S2CID 85911512.
- "International Mouse Phenotyping Consortium".
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A (Jun 2011). "A conditional knockout resource for the genome-wide study of mouse gene function". Nature. 474 (7351): 337–42. doi:10.1038/nature10163. PMC 3572410. PMID 21677750.
- Dolgin E (Jun 2011). "Mouse library set to be knockout". Nature. 474 (7351): 262–3. doi:10.1038/474262a. PMID 21677718. S2CID 39281705.
- Collins FS, Rossant J, Wurst W (Jan 2007). "A mouse for all reasons". Cell. 128 (1): 9–13. doi:10.1016/j.cell.2006.12.018. PMID 17218247. S2CID 18872015.
- White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, et al. (Jul 2013). "Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes". Cell. 154 (2): 452–64. doi:10.1016/j.cell.2013.06.022. PMC 3717207. PMID 23870131.
- "Infection and Immunity Immunophenotyping (3i) Consortium". Archived from the original on 2015-05-21. Retrieved 2015-05-19.
Further reading
- Drüeke TB, Massy ZA (2003). "Advanced oxidation protein products, parathyroid hormone and vascular calcification in uremia". Blood Purification. 20 (5): 494–7. doi:10.1159/000065203. PMID 12207101. S2CID 46752152.
- Parfitt AM (Oct 2002). "Parathyroid hormone and periosteal bone expansion". Journal of Bone and Mineral Research. 17 (10): 1741–3. doi:10.1359/jbmr.2002.17.10.1741. PMID 12369776. S2CID 37111637.
- Martin TJ (Mar 2004). "Does bone reabsorption inhibition affect the anabolic response to parathyroid hormone?". Trends in Endocrinology and Metabolism. 15 (2): 49–50. doi:10.1016/j.tem.2004.01.002. PMID 15080150. S2CID 35482527.
- Keutmann HT, Sauer MM, Hendy GN, O'Riordan LH, Potts JT (Dec 1978). "Complete amino acid sequence of human parathyroid hormone". Biochemistry. 17 (26): 5723–9. doi:10.1021/bi00619a019. PMID 728431.
- Keutmann HT, Niall HD, O'Riordan JL, Potts JT (May 1975). "A reinvestigation of the amino-terminal sequence of human parathyroid hormone". Biochemistry. 14 (9): 1842–7. doi:10.1021/bi00680a006. PMID 1125201.
- Parkinson DB, Thakker RV (May 1992). "A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism". Nature Genetics. 1 (2): 149–52. doi:10.1038/ng0592-149. PMID 1302009. S2CID 24032313.
- Handt O, Reis A, Schmidtke J (Nov 1992). "Ectopic transcription of the parathyroid hormone gene in lymphocytes, lymphoblastoid cells and tumour tissue". The Journal of Endocrinology. 135 (2): 249–56. doi:10.1677/joe.0.1350249. PMID 1474331.
- Tonoki H, Narahara K, Matsumoto T, Niikawa N (1991). "Regional mapping of the parathyroid hormone gene (PTH) by cytogenetic and molecular studies". Cytogenetics and Cell Genetics. 56 (2): 103–4. doi:10.1159/000133059. PMID 1672845.
- Marx UC, Adermann K, Bayer P, Meyer M, Forssmann WG, Rösch P (Feb 1998). "Structure-activity relation of NH2-terminal human parathyroid hormone fragments". The Journal of Biological Chemistry. 273 (8): 4308–16. doi:10.1074/jbc.273.8.4308. PMID 9468478. S2CID 1009667.
- Arnold A, Horst SA, Gardella TJ, Baba H, Levine MA, Kronenberg HM (Oct 1990). "Mutation of the signal peptide-encoding region of the preproparathyroid hormone gene in familial isolated hypoparathyroidism". The Journal of Clinical Investigation. 86 (4): 1084–7. doi:10.1172/JCI114811. PMC 296835. PMID 2212001.
- Nussbaum SR, Gaz RD, Arnold A (Nov 1990). "Hypercalcemia and ectopic secretion of parathyroid hormone by an ovarian carcinoma with rearrangement of the gene for parathyroid hormone". The New England Journal of Medicine. 323 (19): 1324–8. doi:10.1056/NEJM199011083231907. PMID 2215618.
- Ahn TG, Antonarakis SE, Kronenberg HM, Igarashi T, Levine MA (Mar 1986). "Familial isolated hypoparathyroidism: a molecular genetic analysis of 8 families with 23 affected persons". Medicine. 65 (2): 73–81. doi:10.1097/00005792-198603000-00001. PMID 3005800. S2CID 25332134.
- Tregear GW, van Rietschoten J, Greene E, Niall HD, Keutmann HT, Parsons JA, O'Riordan JL, Potts JT (Apr 1974). "Solid-phase synthesis of the biologically active N-terminal 1 - 34 peptide of human parathyroid hormone". Hoppe-Seyler's Zeitschrift für Physiologische Chemie. 355 (4): 415–21. doi:10.1515/bchm2.1974.355.1.415. PMID 4474131. S2CID 43509130.
- Niall HD, Sauer RT, Jacobs JW, Keutmann HT, Segre GV, O'Riordan JL, Aurbach GD, Potts JT (Feb 1974). "The amino-acid sequence of the amino-terminal 37 residues of human parathyroid hormone". Proceedings of the National Academy of Sciences of the United States of America. 71 (2): 384–8. Bibcode:1974PNAS...71..384N. doi:10.1073/pnas.71.2.384. PMC 388010. PMID 4521809.
- Andreatta RH, Hartmann A, Jöhl A, Kamber B, Maier R, Riniker B, Rittel W, Sieber P (1973). "[Synthesis of sequence 1-34 of human parathyroid hormone]". Helvetica Chimica Acta. 56 (1): 470–3. doi:10.1002/hlca.19730560139. PMID 4721748.
- Jacobs JW, Kemper B, Niall HD, Habener JF, Potts JT (May 1974). "Structural analysis of human proparathyroid hormone by a new microsequencing approach". Nature. 249 (453): 155–7. Bibcode:1974Natur.249..155J. doi:10.1038/249155a0. PMID 4833516. S2CID 4226663.
- Vasicek TJ, McDevitt BE, Freeman MW, Fennick BJ, Hendy GN, Potts JT, Rich A, Kronenberg HM (Apr 1983). "Nucleotide sequence of the human parathyroid hormone gene". Proceedings of the National Academy of Sciences of the United States of America. 80 (8): 2127–31. Bibcode:1983PNAS...80.2127V. doi:10.1073/pnas.80.8.2127. PMC 393770. PMID 6220408.
- Mayer H, Breyel E, Bostock C, Schmidtke J (1983). "Assignment of the human parathyroid hormone gene to chromosome 11". Human Genetics. 64 (3): 283–5. doi:10.1007/BF00279412. PMID 6885073. S2CID 35197648.
- Hendy GN, Kronenberg HM, Potts JT, Rich A (Dec 1981). "Nucleotide sequence of cloned cDNAs encoding human preproparathyroid hormone". Proceedings of the National Academy of Sciences of the United States of America. 78 (12): 7365–9. Bibcode:1981PNAS...78.7365H. doi:10.1073/pnas.78.12.7365. PMC 349267. PMID 6950381.
- Hendy GN, Bennett HP, Gibbs BF, Lazure C, Day R, Seidah NG (Apr 1995). "Proparathyroid hormone is preferentially cleaved to parathyroid hormone by the prohormone convertase furin. A mass spectrometric study". The Journal of Biological Chemistry. 270 (16): 9517–25. doi:10.1074/jbc.270.16.9517. PMID 7721880. S2CID 10879253.
External links
 Media related to Parathyroid hormone at Wikimedia Commons
Media related to Parathyroid hormone at Wikimedia Commons- Parathyroid hormone: analyte monograph - the Association for Clinical Biochemistry and Laboratory Medicine
- Overview of all the structural information available in the PDB for UniProt: P01270 (Parathyroid hormone) at the PDBe-KB.
PDB gallery | |
|---|---|
|