Spiradenoma
Spiradenomas (SA) are rare, benign cutaneous adnexal tumors that may progress to become their malignant counterparts, i.e. spiradenocarcinomas (SAC). Cutaneous adnexal tumors are a group of skin tumors consisting of tissues that have differentiated (i.e. matured from stem cells) towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands.[1] SA and SAC tumors were regarded as eccrine gland tumors and termed eccrine spiradenomas and eccrine spiradenocarcinomas, respectively. However, more recent studies have found them to be hair follicle tumors[2] and commonly term them spiradenomas and spiradenocarcinomas, respectively.[2][3] Further confusing the situation, SA-like and SAC-like tumors are also 1) manifestations of the inherited disorder, CYLD cutaneous syndrome (CCS), and 2) have repeatedly been confused with an entirely different tumor, adenoid cystic carcinomas of the salivary gland.[4] Here, SA and SAC are strictly defined as sporadic hair follicle tumors that do not include the hereditary CCS spiradenomas and heridtary spiradenocarcinoms of CCS or the adenoid cystic carcinomas.
| Spiradenomas | |
|---|---|
| Other names | Eccrine spiradenoma |
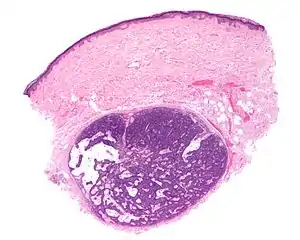 | |
| Micrograph of a spiradenoma (bottom-center of image). H&E stain. | |
| Specialty | Dermatology, Surgery, Oncology |
| Symptoms | Solitary skin tumor |
| Risk factors | development of malignant tumors |
SA tumors usually occur as slow-growing,[5] single, small, nodular lesions located in the skin of the head, neck, or trunk.[3] SAC tumors develop from benign SA tumors or in very rare cases begin as malignant tumors.[6] SA and SAC tumors must be distinguished from the spiradenoma and spiradenocaricnoma tumors that develop in individuals afflicted with the CYLD cutaneous syndrome. CCS is an inherited disorder that commonly involves the development of multiple but on occasion a single or few tumors that closely resemble, and may be confused with, the sporadic SA and SAC tumors described here.[4][7][8] CCS spiradenoma and CCS spiradenocarcinoma tumors must be distinguished form the sporadic SA and SAC tumors reviewed here in order to afford genetic counseling to individuals with CCS as well as to the close family members of these individuals.[9]
Currently, SA[3] and SAC[5] are usually treated by complete excision making sure that no tumor cells are left behind at the surgical site. This is particularly important in SAC where the incomplete surgical removal of all tumor cells may result in recurrence of the tumor at the surgical site and/or its metastasis to the local lymph nodes draining the surgical site and/or to distant tissues.[5] In addition to surgical resection, some cases of SAC tumors have been treated with adjuvant therapy that includes radiotherapy and/or chemotherapy.[6] It is not clear that these adjuvant treatments improve patient prognoses. Further studies are needed to determine the best treatment(s) for localize SA, localized SAC, and, in particular, metastatic SAC tumors.[5]
Presentation
Spiradenoma tumors occur in individuals of various ages but tend to develop in middle-aged and elder adults (e.g. in a study of 27 patients, the median age at diagnosis was 62 years).[2] However, cases have been reported in children as young as 2 years.[10] These tumors usually present as a solitary, sometimes painful,[11] slowly growing, 1–2 cm (i.e. centimeters), gray-to-pink nodules that lie underneath the skin's epidermis in the head, neck, trunk, arms, or legs.[12] However, these nodules have been reported to be as large as 6 cm[11] and to occur in other hair follicle-containing cutaneious sites such as the vulva,[13] breast, and breast nipple.[14] Areas of the skin that do not have hair follicles (e.g. palms of the hands and soles of feet) do not develop these nodules.[11] Less than 2% of cases present with more than one nodule; the multiple nodules in SA tend to array in a linear, blaschkoid (i.e. V-, U-, or inverted U-shaped), or zosteriform (i.e. belt- or girdle-shape tracking an area of skin served by a sensory nerve) patterns.[12] Giant vascular eccrine spiradenomas are larger (> 2 cm) and more highly vascular variants of SA that usually develop at the same cutaneous sites as SA but have also been reported to occur in the abdomen and scrotum. No malignant giant vascular SA has been described in the literature to date (2021).[12]
A recent review of 182 cases reported that spiradenocarcinomas were diagnosed in individuals 8–89 years old (mean 57.4 years). These SAC tumors developed in SA tumors that had existed for 0–720 months (mean 168.5 months); 14.4% of individuals were diagnosed with multiple carcinoma lesions. (Multiple SAC lesions may appear as a nodule associated with smaller satellite nodules.[6]) The lesions occurred on the trunk (32.3% of cases), limbs (31.3%), head or neck (30.7%), and genitalia (1.6%). These individuals presented with lesions that began to show abnormal growth (82.6% of cases), pain (47.7% of cases), and/or ulcerations (38.7% of cases).[5] In earlier studies, SAC tumors were reported to varied in size with 40% being larger than 5 cm and the majority not having metastasized; the sites to which these lesions metastasized were most commonly to lymph nodes near to the SAC or, rarely, distant tissues such as the lung, liver, brain, spinal cord, bone, skin, and breast.[6][15][16]
Pathology
As determined by the microscopic histopathological appearances of their hematoxylin and eosin-stained samples, spiradenomas are non-encapsulated nodular skin lesions that extend into the dermis. The lesions consist of a relatively disorganized and dense array of proliferating basophilic cells (i.e. cells appearing blue because of their abnormally large uptake of the hematoxylin stain).[9] These cells are arranged as small cells with hyperchromatic nuclei and scant cytoplasm layered outside and larger cells with vesicular (i.e. vesicle-containing) nuclei layered inside of intertwining strips.[16] Lymphocytes commonly populate these lesions. Some lesions may merge the histopathological features of spiroadenomas with those of cylindromas.[9] Cylindromas are hair follicle tumors that consist of basal cells (i.e. small, round cells similar to those seen in the lowest layer of the skin's epidermis) which are arranged in ("jigsaw-like"[17] cylindrical patterns separated by thickened basement membranes. The cylindroma tissues may also contain disorganized, dense arrays of proliferating basophilic cells.[9]
Spiradenocarcinoma tumors almost always derive from SA tumors[18] and have areas of SA architecture abruptly transitioning to areas of SAC architecture. The SAC component of these lesions consists of cells that, along with their nuclei and cytoplasm, vary greatly in size and shape. The cells may be rapidly proliferating, have atypical mitosis figures, and may be invading nearby neural or lymphovascular structures; they do not arrange into intertwining strips of small and large cells.[16] High-grade SAC tissues show a more severely disturbed architecture, more marked variation in cell, nuclei, and cytoplasm sizes and shapes, and more rapidly proliferating cells than low-grade SAC tissues.[19] SAC tumor tissues may show various other features, e.g. they may closely resemble sarcomas.[19] In rare cases, SAC has been reported to apparently arise de novo, i.e. begin as malignant lesions.[18]
Gene abnormalities
Studies indicate that a minority of SA and SAC cases have one of two potentially disease-causing mutations in their tumor cells: loss-of-function (i.e. gene-inactivating) mutations in the CYLD gene in 5 of 17 SA and 2 of 24 SAC cases or gain-of-function (i.e. gene-activating) mutations in the ALPK1 gene in 7 of 16 SA and 4 of 14 SAC cases. The two mutations are mutually exclusive, i.e. do not occur together in any individual's tumor(s).[20] Both mutations result in blocking the actions pf NF-κB.[20][17] NF-κB is a transcription factor which acts indirectly to stimulate the expression of various genes that trigger cell death by apoptosis and necroptosis[21] and also inhibit cell proliferation (see CYLD gene's mechanism of action).[22] It is proposed that the blockade of NF-κB's actions prolongs cell survival, stimulates high rates of cell growth, and perhaps has other effects which contribute to the formation and/or progression of SA and SAC tumors.[9][23]
Diagnosis
SA and SAC tumors have been confused with leiomyoma, angiolipoma, granular cell tumor,[11] basal cell carcinoma, squamous cell carcinoma, metastatic adenocarcinoma, and adenoid cystic carcinoma tumors;[19] the giant vascular SA form of these tumors has been mistaken for a hemangioma, angiolipoma, calcifying epithelioma, neuroma, malignant melanoma, angiosarcoma, vascular malformation, and venous thrombosis.[12] CT scans, FDG-positron emission tomography, and magnetic resonance imaging to detect 1) the shape and local invasiveness of these tumors and 2) the presence of metastases[5] and 2) the clinical presentation of individuals of these tumors often suggest the correct diagnosis. However, definitive separation of SA, SAC, and giant vascular SA from the just cited lesions requires histopathological examinations.[11][17] The diagnosis of SA versus SAC tumors is often suggested by their clinical presentations, e.g. rapid enlargement or onset of pain in a long-standing SA tumor strongly suggests that it has become a SAC.[16] Findings that a known SA or undiagnosed tumor has areas of SA histology transitioning to SAC histology (see above Pathology section) is virtually diagnostic for SAC.[19]
The sporadic SA and SAC tumors reviewed here must also be distinguished from the very similar appearing SA-like and SAC-like tumors in individuals afflicted with the CYLD cutaneous syndrome. CCS is a familial disease uniformly associated with the inheritance of inactivating mutations in the CYLD gene. Individuals with CCS commonly develop an increasingly large number of skin tumors, including SA and SAC, over time. Usually, these individuals are readily distinguished from sporadic SA and SAC by their family history of the disease and the presence of large numbers of tumors. The rare cases of CCS that present with one or a few tumors are distinguished form CCS by testing for the CYLD gene: all individuals with CCS carry a mutated CYLD gene in their tumor as well as other tissues such as blood leucocytes while individuals with sporadic SA or SAC carry the mutated CYLD gene in their tumor but not other tissues (see Genetics of CCS). It is important to afford individuals with CCS and the close family members of these individuals access to in depth genetic counselling.[9]
Treatments
There is no consensus on the best treatment(s) for SA and SAC tumors. Further research, including meta-analysis comparisons of different treatment regimens, is needed to define the best treatment(s) for these tumors. In all cases, however, regular follow-ups of individuals with SA and SAC are strongly recommended in order to detect as early as possible: the formation of new tumors; changes in old tumors that suggest their progression to more aggressive or malignant formss; local recurrences of surgically removed tumors; and the development of local or distant metastases.[5][11] One recommended follow-up strategy prescribes patients to have check-ups ever three months in the first year after treatment, every six months in the second year after treatment, and annually thereafter. Each check-up should incorporate the examination of the patients' regional lymph nodes and on a case-by-case basis annual chest X-rays and liver function tests.[5]
Spiradenomas
The currently preferred treatment for sporadic SA tumors (including giant vascular eccrine SA) in almost all cases is surgical excision.[5] Many studies recommend wide local surgical excision of these tumors with surgical margins of ≥1 cm in order to ensure that all tumor cells are removed: local lymph node resections are usually reserved for those cases suspected of having lymph node metastases.[5][6][12] Mohs micrographic surgery has been used as an alternative to surgical excision for the complete removal of these tumors.[6] Since SA tumors may contain unidentified areas of malignancy and therefore have high recurrence rates, may metastasize to local lymph nodes and/or distant tissues, and can result in overall poor survival times,[16] adjuvant radiation therapy has been used to minimize these aggressive events in SA tumors with equivocal signs of malignancy. However, the effectiveness of these adjuvant therapies has not been established.[6] Adjuvant CO2 laser treatments have been used on occasion to remove these tumors in cases with multiple tumors.[3] Some trials have using botulinum toxin A and triamcinolone injections directly into SA tumors[10] but these trials have had limited success.[11]
Spiradenocarcinomas
With only 117 malignant cases reported in the literature as of 2022, there are no standard guidelines for the treatment of SAC tumors.[24] Localized (i.e. no evidence of metastases) SAC tumors and SAC tumors that have metastasized to local lymph nodes are commonly treated by surgical removal (with ≥1 cm margins) of the tumor plus surgical resection (i.e. removal) of the local lymph nodes. Individuals presenting with lymph node metastasis have also been treated with one or both of theses regimens plus, where possible and indicated, adjuvant radiation therapy to the areas of the removed tumors and lymph nodes.[5] Chemotherapeutic agents such as methotrexate, cisplatin, etoposide, and 5-fluorouracil have been used to treat SAC with distant tissue metastases but the success of these drug treatments has been limited.[6]
Prognoses
In the only meta-analysis study reported on the treatment of SA and SAC tumors to date, 35 individuals with SA tumors and no evidence of local or distant metastasis who were treated with local resection (3 of these individuals underwent additional lymph node resections after removal of their primary tumor) all 35 were disease-free with a mean follow-up period of 33 months. Among individuals with SAC tumors that had metastasized to lymph nodes but not distant tissues, 3 individuals who had surgical excisions of their tumors but not their involved lymph nodes died of metastatic disease (range 39–49 months, mean survival range 45 months); one individual was alive with disease 24 months after lymph node dissection; and 8 individuals (6 of whom had lymph node dissection) lacked evidence of recurrent disease (observation times 2 to 97 months, mean observation time 47 months).[6] In a review of 136 reported cases treated for SA or SAC: 1) 4.4% and 14.7% of individuals with SA and SAC tumors, respectively, had died (time between treatment and death was not reported); 2) patients who presented with metastasis to distant tissues had a median survival time of 16 months (11 of these patients were treated with adjuvant therapy in addition to surgery [adjuvant treatments included 5 patients treated with radiotherapy, 3 treated with chemotherapy, and 2 treated with both radiotherapy and chemotherapy]); and 3) Surgery with adjuvant therapy yielded a median survival time of 20 months (this survival time did not significantly differ from that found in individuals treated with surgery alone).[5]
See also
References
- Zaballos P, Gómez-Martín I, Martin JM, Bañuls J (October 2018). "Dermoscopy of Adnexal Tumors". Dermatologic Clinics. 36 (4): 397–412. doi:10.1016/j.det.2018.05.007. PMID 30201149. S2CID 52185272.
- Sellheyer K (February 2015). "Spiradenoma and cylindroma originate from the hair follicle bulge and not from the eccrine sweat gland: an immunohistochemical study with CD200 and other stem cell markers". Journal of Cutaneous Pathology. 42 (2): 90–101. doi:10.1111/cup.12406. PMID 25354097. S2CID 25569722.
- Płachta I, Kleibert M, Czarnecka AM, Spałek M, Szumera-Ciećkiewicz A, Rutkowski P (May 2021). "Current Diagnosis and Treatment Options for Cutaneous Adnexal Neoplasms with Apocrine and Eccrine Differentiation". International Journal of Molecular Sciences. 22 (10): 5077. doi:10.3390/ijms22105077. PMC 8151110. PMID 34064849.
- Macagno N, Sohier P, Kervarrec T, Pissaloux D, Jullie ML, Cribier B, Battistella M (January 2022). "Recent Advances on Immunohistochemistry and Molecular Biology for the Diagnosis of Adnexal Sweat Gland Tumors". Cancers. 14 (3): 476. doi:10.3390/cancers14030476. PMC 8833812. PMID 35158743.
- Wagner K, Jassal K, Lee JC, Ban EJ, Cameron R, Serpell J (October 2021). "Challenges in diagnosis and management of a spiradenocarcinoma: a comprehensive literature review". ANZ Journal of Surgery. 91 (10): 1996–2001. doi:10.1111/ans.16626. PMID 33522696. S2CID 231767765.
- Andreoli MT, Itani KM (May 2011). "Malignant eccrine spiradenoma: a meta-analysis of reported cases". American Journal of Surgery. 201 (5): 695–9. doi:10.1016/j.amjsurg.2010.04.015. PMID 20851376.
- Chauhan DS, Guruprasad Y (January 2012). "Dermal cylindroma of the scalp". National Journal of Maxillofacial Surgery. 3 (1): 59–61. doi:10.4103/0975-5950.102163. PMC 3513812. PMID 23251061.
- Kazakov DV, Zelger B, Rütten A, Vazmitel M, Spagnolo DV, Kacerovska D, Vanecek T, Grossmann P, Sima R, Grayson W, Calonje E, Koren J, Mukensnabl P, Danis D, Michal M (May 2009). "Morphologic diversity of malignant neoplasms arising in preexisting spiradenoma, cylindroma, and spiradenocylindroma based on the study of 24 cases, sporadic or occurring in the setting of Brooke-Spiegler syndrome". The American Journal of Surgical Pathology. 33 (5): 705–19. doi:10.1097/PAS.0b013e3181966762. PMID 19194280. S2CID 9858971.
- Nagy N, Dubois A, Szell M, Rajan N (2021). "Genetic Testing in CYLD Cutaneous Syndrome: An Update". The Application of Clinical Genetics. 14: 427–444. doi:10.2147/TACG.S288274. PMC 8566010. PMID 34744449.
- Gordon S, Styron BT, Haggstrom A (2013). "Pediatric segmental eccrine spiradenomas: a case report and review of the literature". Pediatric Dermatology. 30 (6): e285–6. doi:10.1111/j.1525-1470.2012.01777.x. PMID 22612572. S2CID 46430687.
- Ren F, Hu Z, Kong Q, Sang H (August 2015). "Multiple Segmental Eccrine Spiradenoma with a Zosteriform Pattern: A Case Report and Literature Review". Annals of Dermatology. 27 (4): 435–8. doi:10.5021/ad.2015.27.4.435. PMC 4530155. PMID 26273161.
- Li Z, Li G, Jiang X, Fu X (April 2021). "Giant vascular eccrine spiradenoma: the first case in the scrotum and review of the literature author". Diagnostic Pathology. 16 (1): 37. doi:10.1186/s13000-021-01073-8. PMC 8091673. PMID 33941210.
- Heller DS (September 2015). "Benign Tumors and Tumor-like Lesions of the Vulva". Clinical Obstetrics and Gynecology. 58 (3): 526–35. doi:10.1097/GRF.0000000000000133. PMID 26125957. S2CID 11112420.
- Metovic J, Gallino C, Zanon E, Bussone R, Russo R, Vissio E, Annaratone L, Conti L, Papotti M, Cassoni P, Castellano I (August 2019). "Eccrine spiradenoma of the nipple: Case report, differential diagnosis and literature review". Histology and Histopathology. 34 (8): 909–915. doi:10.14670/HH-18-094. PMID 30806477.
- Avraham JB, Villines D, Maker VK, August C, Maker AV (July 2013). "Survival after resection of cutaneous adnexal carcinomas with eccrine differentiation: risk factors and trends in outcomes". Journal of Surgical Oncology. 108 (1): 57–62. doi:10.1002/jso.23346. PMID 23677677. S2CID 27443162.
- Huang A, Vyas NS, Mercer SE, Phelps RG (April 2019). "Histological findings and pathologic diagnosis of spiradenocarcinoma: A case series and review of the literature". Journal of Cutaneous Pathology. 46 (4): 243–250. doi:10.1111/cup.13408. PMID 30588645. S2CID 58592021.
- Plotzke JM, Adams DJ, Harms PW (January 2022). "Molecular pathology of skin adnexal tumours". Histopathology. 80 (1): 166–183. doi:10.1111/his.14441. PMID 34197659. S2CID 235714739.
- Staiger RD, Helmchen B, Papet C, Mattiello D, Zingg U (October 2017). "Spiradenocarcinoma: A Comprehensive Data Review" (PDF). The American Journal of Dermatopathology. 39 (10): 715–725. doi:10.1097/DAD.0000000000000910. PMID 28570391. S2CID 41154006.
- Held L, Ruetten A, Saggini A, Kempter W, Tiedke C, Weber-Kuhn S, De Saint Aubain N, Mentzel T (March 2021). "Metaplastic spiradenocarcinoma: Report of two cases with sarcomatous differentiation". Journal of Cutaneous Pathology. 48 (3): 384–389. doi:10.1111/cup.13897. PMID 33051901. S2CID 222352535.
- Rashid M, van der Horst M, Mentzel T, Butera F, Ferreira I, Pance A, Rütten A, Luzar B, Marusic Z, de Saint Aubain N, Ko JS, Billings SD, Chen S, Abi Daoud M, Hewinson J, Louzada S, Harms PW, Cerretelli G, Robles-Espinoza CD, Patel RM, van der Weyden L, Bakal C, Hornick JL, Arends MJ, Brenn T, Adams DJ (May 2019). "ALPK1 hotspot mutation as a driver of human spiradenoma and spiradenocarcinoma". Nature Communications. 10 (1): 2213. Bibcode:2019NatCo..10.2213R. doi:10.1038/s41467-019-09979-0. PMC 6525246. PMID 31101826.
- Lork M, Verhelst K, Beyaert R (July 2017). "CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different". Cell Death and Differentiation. 24 (7): 1172–1183. doi:10.1038/cdd.2017.46. PMC 5520167. PMID 28362430.
- Vlahopoulos SA (August 2017). "Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode". Cancer Biology & Medicine. 14 (3): 254–270. doi:10.20892/j.issn.2095-3941.2017.0029. PMC 5570602. PMID 28884042.
- Cui Z, Kang H, Grandis JR, Johnson DE (January 2021). "CYLD Alterations in the Tumorigenesis and Progression of Human Papillomavirus-Associated Head and Neck Cancers". Molecular Cancer Research. 19 (1): 14–24. doi:10.1158/1541-7786.MCR-20-0565. PMC 7840145. PMID 32883697.
- Wargo JJ, Carr DR, Plaza JA, Verschraegen CF (February 2022). "Metastatic Spiradenocarcinoma Managed With PD-1 Inhibition". Journal of the National Comprehensive Cancer Network. 20 (4): 318–320. doi:10.6004/jnccn.2021.7119. PMID 35196645. S2CID 247082711.