Alport syndrome
Alport syndrome is a genetic disorder[1] affecting around 1 in 5,000-10,000 children,[2] characterized by glomerulonephritis, end-stage kidney disease, and hearing loss.[3] Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.[4]
| Alport syndrome | |
|---|---|
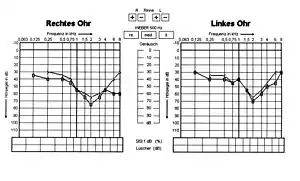 | |
| Hearing loss effect of Alport syndrome in 13-year-old boy | |
| Specialty | Medical genetics |
The disorder was first identified in a British family by the physician Cecil A. Alport in 1927.[5][6] Alport syndrome once also had the label hereditary nephritis, but this is misleading as there are many other causes of hereditary kidney disease and 'nephritis'.
Alport syndrome is caused by an inherited defect in type IV collagen—a structural material that is needed for the normal function of different parts of the body. Since type IV collagen is found in the ears, eyes, and kidneys, this explains why Alport syndrome affects different seemingly unrelated parts of the body (ears, eyes, kidneys, etc.).
Depending on where the mutation is located in the genome, Alport syndrome can present itself in many forms. This includes X-linked Alport syndrome (XLAS), autosomal recessive Alport syndrome (ARAS), and autosomal dominant Alport syndrome (ADAS).[7]
Signs and symptoms
These descriptions refer to 'classic' Alport syndrome, which usually causes significant disease from young adult or late childhood life.[8] Some individuals, usually with milder mutations or 'carrier' status, develop disease later, or show only some of the features of classic disease.
Chronic kidney disease
Blood in urine is a usual feature of Alport syndrome from early infancy, identifiable on urine dipsticks. In young children, episodes of visible (macroscopic) haematuria may occur. Protein begins to appear in urine as the disease progresses. This is now regarded as an indication for treatment with ACE inhibitors. Progressive loss of kidney function (reflected clinically by increases in serum creatinine or decreases in estimated glomerular filtration rate) can occur and may require treatment with renal replacement: dialysis or a kidney transplant.[9]
Hearing loss
Alport syndrome can also cause hearing loss although some patients are not affected.[10] Hearing in Alport syndrome patients is normal at birth. Hearing loss in affected patients develops progressively, usually at the stage when kidney function is normal, but there is substantial proteinuria. However, in some patients, hearing loss is only noted after kidney function has been lost. Characteristically the early changes are reduced ability to hear high-frequency sounds, 'sensorineural deafness'. This becomes more severe and affects lower frequencies too. Hearing loss is not usually complete in Alport syndrome; good communication is almost always possible with the use of hearing aids.[11]
Eye changes
Various eye abnormalities are often seen including lenticonus, keratoconus, cataracts and corneal erosion as well as retinal flecks in the macula and mid-periphery.[12] These rarely threaten vision. Lenticonus (cone-shaped lens) can be treated by replacement of the lens, as for cataracts. Mild keratoconus can be managed with hard, scleral, piggy-back or other specialty medical contact lenses; progressive cases may be halted with corneal collagen cross linking; and severe cases may require a corneal transplant. Macular abnormalities such as incomplete foveal hypoplasia or staircase foveopathy are common in Alport syndrome.[13]
It may also be associated with retinitis pigmentosa.[14]
Leiomyomatosis
Diffuse leiomyomatosis of the oesophagus and tracheobronchial tree has been reported in some families with Alport syndrome. Symptoms usually appear in late childhood and include dysphagia, postprandial vomiting, substernal or epigastric pain, recurrent bronchitis, dyspnea, cough, and stridor. Leiomyomatosis is confirmed by computed tomography (CT) scanning or magnetic resonance imaging (MRI).[15]
Other abnormalities
Aortic dissection has been described very rarely in patients with early-onset disease.[8] Leiomyomas, tumours of smooth muscle affecting the oesophagus and female genital tract, may occur in a rare overlap syndrome involving the adjacent COL4A5 and COL4A6 genes.[16]
Pathophysiology
Alport Syndrome is a relatively common genetic disorder affecting around 1 in 5,000-10,000 children,[2]
Genetics
Alport syndrome is caused by mutations in COL4A3, COL4A4, and COL4A5, three of six human genes involved in basement membrane (type IV) collagen biosynthesis. Mutations in any of these genes prevent the proper production or assembly of the specialised type IV collagen '345' network which is an important structural component of basement membranes in the kidney, inner ear, and eye.[17] It is also found in other locations, including the alveoli of the lungs. Basement membranes are thin, sheet-like structures that separate and support cells in many tissues. Type IV collagen '112' type is found in both vertebrates and invertebrates and is the major isoform in most human basement membranes. When mutations prevent the formation of 345 type IV collagen network in the glomerulus, the 112 network, which is formed in fetal development but usually replaced by 345, persists into adult life.[18]
Inheritance patterns
Alport syndrome can have different inheritance patterns depending on which specific mutation is present.
- In most people with Alport syndrome (about 85%), the condition is inherited in an X-linked pattern,[19] due to mutations in the COL4A5 gene. A condition is considered X-linked if the gene involved in the disorder is located on the X chromosome. In males, who have only one X chromosome, one altered copy of the COL4A5 gene is sufficient to cause severe Alport syndrome, explaining why most affected males eventually develop kidney failure. In females, who have two X chromosomes, a mutation in one copy of the COL4A5 gene usually results in blood in the urine, but most affected females do not develop kidney failure.
- Alport syndrome can also be inherited in an autosomal recessive pattern if both copies of the COL4A3 or COL4A4 gene, located on chromosome 2, have been mutated.[17] Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.[9]
- Past descriptions of an autosomal dominant form are now usually categorized as other conditions.[20] Notably, conditions associated with giant platelets and associated with mutations of MYH9 are no longer considered to be Alport variants. However apparent autosomal dominant transmission of disease associated with mutations in COL4A3 and COL4A4 does occur.[21][22]
Clinical utility gene card for: Alport syndrome.[23]
Diagnosis
The diagnosis can usually be made on a combination of clinical, family history, and biopsy criteria.
Biopsy of kidneys or skin
To be helpful, kidney biopsies need to be taken before the disease is too advanced. Changes on conventional (light) microscopy are not characteristic, and the possibility of other diagnoses, particularly focal segmental glomerulosclerosis (FSGS), may be raised. Electron microscopy shows a characteristic sequence of changes from thinning of the glomerular basement membrane (GBM), developing into areas of thinning and thickening, and finally into a complex appearance with apparent splitting, often described as a 'basketweave' appearance. Early or very localised changes on this spectrum are not diagnostic, but the later changes are considered diagnostic.
Immunohistochemistry or immunofluorescence studies to identify the COL3-4-5 proteins in GBM can be helpful. However, these studies may be normal in some patients with Alport syndrome, especially milder variants.
The skin contains type IV collagen in a '556' network. Skin biopsies have been used to show the absence of the COL4A5 gene product, but these techniques are not straightforward, only apply to patients with severe COL4A5 mutations, and are not widely available. Genetic testing is now a better alternative if kidney biopsy is not possible.
Family history
A family history of end-stage renal disease with hearing impairment is suggestive of Alport syndrome, but other conditions can cause this combination of abnormalities. Most can be distinguished by clinical features. The finding of haematuria in relatives is suggestive. While X-linked inheritance is the most common pattern, genetic testing is revealing that atypical presentations may be more common than currently thought.
Genetic testing
Genetic testing plays an increasingly important role in confirming the diagnosis where the clinical features do not amount to proof.[24]
Treatment
Kidney disease and renal failure
In addition to measures for chronic kidney disease (CKD) of any cause, there is evidence that ACE inhibitors can slow the deterioration of kidney function in Alport syndrome, delaying the need for dialysis or transplantation.[26] The development of proteinuria has been recommended as an indication for commencing treatment.[8]
Once kidney failure has developed, patients usually do well on dialysis or with a kidney transplant. Transplantation can rarely be associated with the formation of antibodies to type IV collagen in the donor kidney resulting in progressive graft failure as a result of Goodpasture syndrome ('Alport post-transplant anti-GBM disease').[27][28]
Gene therapy has been frequently discussed, but delivering it to the podocytes in the glomerulus that normally produce the type IV collagen in the glomerular basement membrane is challenging.[29]
Hearing loss
It is not known whether ACE inhibitors or other treatments affect hearing loss. For those with classic Alport syndrome, hearing aids are often required in teenage or young adult years.[30]
Prognosis
Studies of the life expectancy of patients with Alport syndrome are rare, but one 2012 study found that Alport patients receiving renal replacement therapy (dialysis or kidney transplantation) exhibited, on average, better survival compared with matched controls who had other renal diseases (and who also received renal replacement therapy).[31]
See also
- AMMECR1
- Samoyed hereditary glomerulopathy, a disease shown to be a model for Alport syndrome.[32]
- Fechtner syndrome
- Thin basement membrane disease
References
- "Diseases of the Kidney: Alport Syndrome". Archived from the original on 2004-06-12. Retrieved 2004-06-16.
- "What is Alport syndrome?". Alport syndrome. Archived from the original on 2019-01-06. Retrieved 2019-01-06.
- "Alport syndrome" at Dorland's Medical Dictionary
- "Alport Syndrome". National Kidney Foundation. 2015-12-24. Retrieved 2022-08-01.
- Lagona E, Tsartsali L, Kostaridou S, Skiathitou A, Georgaki E, Sotsiou F (April 2008). "Skin biopsy for the diagnosis of Alport syndrome". Hippokratia. 12 (2): 116–8. PMC 2464308. PMID 18923659.
- Alport AC (March 1927). "Hereditary familial congenital haemorrhagic nephritis". British Medical Journal. 1 (3454): 504–6. doi:10.1136/bmj.1.3454.504. JSTOR 25322864. PMC 2454341. PMID 20773074.
- Zaunbrecher, Nicole. "Types of Alport Syndrome". Alport Syndrome News. Retrieved 2022-08-01.
- UK Alport Group (2013-07-25). "Alport SyndromeL Clinician information". RareRenal. Renal Rare Diseases Registry. Retrieved 17 February 2016.
- "Alport syndrome". MedlinePlus. Retrieved 28 June 2021.
- Zhou J, Hertz JM, Tryggvason K (June 1992). "Mutation in the alpha 5(IV) collagen chain in juvenile-onset Alport syndrome without hearing loss or ocular lesions: detection by denaturing gradient gel electrophoresis of a PCR product". American Journal of Human Genetics. 50 (6): 1291–300. PMC 1682577. PMID 1598909.
- Watson S, Padala SA, Bush JS (28 May 2020). "Alport Syndrome". StatPearls. PMID 29262041. Retrieved 30 May 2020.
{{cite journal}}: Cite journal requires|journal=(help) - Chugh KS, Sakhuja V, Agarwal A, Jha V, Joshi K, Datta BN, et al. (1993). "Hereditary nephritis (Alport's syndrome)--clinical profile and inheritance in 28 kindreds". Nephrology, Dialysis, Transplantation. 8 (8): 690–5. doi:10.1093/ndt/8.8.690. PMID 8414153.
- Hess K, Pfau M, Wintergerst MW, Loeffler KU, Holz FG, Herrmann P (February 2020). "Phenotypic Spectrum of the Foveal Configuration and Foveal Avascular Zone in Patients With Alport Syndrome". Invest Ophthalmol Vis Sci. 61 (2): 5. doi:10.1167/iovs.61.2.5. PMC 7324255. PMID 32031577.
- Columbia University, Department of Ophthalmology. Link https://www.columbiaeye.org/content/retinitis-pigmentosa
- Alport Syndrome~clinical at eMedicine
- Kashtan CE (February 2019). "Alport Syndrome". In Adam MP, Ardinger HH, Pagon RA, et al. (eds.). Gene Reviews. Seattle (WA): University of Washington, Seattle. PMID 20301386.
- Nozu K, Nakanishi K, Abe Y, Udagawa T, Okada S, Okamoto T, et al. (February 2019). "A review of clinical characteristics and genetic backgrounds in Alport syndrome". Clinical and Experimental Nephrology. 23 (2): 158–168. doi:10.1007/s10157-018-1629-4. PMC 6510800. PMID 30128941.
- "Alport Syndrome". The Lecturio Medical Concept Library. Retrieved 28 June 2021.
- Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. (October 2003). "X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study". Journal of the American Society of Nephrology. 14 (10): 2603–10. doi:10.1097/01.ASN.0000090034.71205.74. PMID 14514738.
- "Alport Syndrome, Autosomal Dominant". Online Mendelian Inheritance in Man (OMIM). Johns Hopkins University. Retrieved 2008-11-24.
- Kharrat M, Makni S, Makni K, Kammoun K, Charfeddine K, Azaeiz H, et al. (September 2006). "Autosomal dominant Alport's syndrome: study of a large Tunisian family". Saudi Journal of Kidney Diseases and Transplantation. 17 (3): 320–5. PMID 16970251.
- Pescucci C, Mari F, Longo I, Vogiatzi P, Caselli R, Scala E, et al. (May 2004). "Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene". Kidney International. 65 (5): 1598–603. doi:10.1111/j.1523-1755.2004.00560.x. PMID 15086897.
- Hertz JM, Thomassen M, Storey H, Flinter F (June 2012). "Clinical utility gene card for: Alport syndrome". European Journal of Human Genetics. 20 (6): 713. doi:10.1038/ejhg.2011.237. PMC 3355248. PMID 22166944.
- "Alport Syndrome". The Lecturio Medical Concept Library. Retrieved 8 July 2021.
- Zhang KW, Colville D, Tan R, Jones C, Alexander SI, Fletcher J, Savige J (August 2008). "The use of ocular abnormalities to diagnose X-linked Alport syndrome in children". Pediatric Nephrology. 23 (8): 1245–50. doi:10.1007/s00467-008-0759-4. PMID 18343956. S2CID 28650514.
- Alport Syndrome~treatment at eMedicine
- "Alport syndrome". Renal Unit at the Royal Infirmary of Edinburgh, Scotland.
- "EdRen - Edinburgh Royal Infirmary Renal Unit - Alport anti-GBM disease". www.edren.org. Retrieved 2016-02-17.
- Tryggvason K, Heikkilä P, Pettersson E, Tibell A, Thorner P (May 1997). "Can Alport syndrome be treated by gene therapy?". Kidney International. 51 (5): 1493–9. doi:10.1038/ki.1997.205. PMID 9150464.
- "Alport Syndrome". The Lecturio Medical Concept Library. Retrieved 28 June 2021.
- Temme J, Kramer A, Jager KJ, Lange K, Peters F, Müller GA, et al. (December 2012). "Outcomes of male patients with Alport syndrome undergoing renal replacement therapy". Clinical Journal of the American Society of Nephrology. 7 (12): 1969–76. doi:10.2215/CJN.02190312. PMC 3513741. PMID 22997344.
- Chen D, Jefferson B, Harvey SJ, Zheng K, Gartley CJ, Jacobs RM, Thorner PS (March 2003). "Cyclosporine a slows the progressive renal disease of alport syndrome (X-linked hereditary nephritis): results from a canine model". Journal of the American Society of Nephrology. 14 (3): 690–8. doi:10.1097/01.ASN.0000046964.15831.16. PMID 12595505.
![]() This article incorporates public domain material from Alport syndrome. United States National Library of Medicine. (Genetics Home Reference).
This article incorporates public domain material from Alport syndrome. United States National Library of Medicine. (Genetics Home Reference).