Osteogenesis imperfecta
Osteogenesis imperfecta (IPA: /ˌɒstioʊˈdʒɛnəsɪs ˌɪmpɜːrˈfɛktə/;[4] OI), colloquially known as brittle bone disease, is a group of genetic disorders that all result in bones that break easily.[1]: 85 [9] The range of symptoms—on the skeleton as well as on the body's other organs—may be mild to severe.[5]: 1512 Symptoms found in various types of OI include whites of the eye (sclerae) that are blue instead, short stature, loose joints, hearing loss, breathing problems[10] and problems with the teeth (dentinogenesis imperfecta).[5] Potentially life-threatening complications, all of which become more common in more severe OI, include: tearing (dissection) of the major arteries, such as the aorta;[1]: 333 [11] pulmonary valve insufficiency secondary to distortion of the ribcage;[1]: 335–341 [12] and basilar invagination.[13]: 106–107
| Osteogenesis imperfecta (OI) | |
|---|---|
| Other names | Brittle bone disease,[1] Lobstein syndrome,[1]: 5 fragilitas ossium,[2] Vrolik disease,[1]: 5 osteopsathyrosis idiopathica[3]: 347 |
 | |
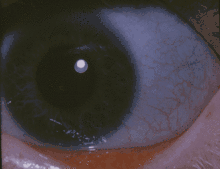
| Blue sclerae, as in the eyes of the girl above, are a classic non-pathognomonic sign of OI. | |
| Pronunciation |
|
| Specialty | Pediatrics, medical genetics, orthopedics |
| Symptoms | Bones that break easily, blue tinge to the sclera (whites of the eye), short height, joint hypermobility, hearing loss[5] |
| Onset | Birth |
| Duration | Long term |
| Causes | Genetic (autosomal dominant or de novo mutation)[6] |
| Diagnostic method | Based on symptoms, DNA testing |
| Prevention | Pre-implantation genetic diagnosis |
| Management | Healthy lifestyle (exercise, no smoking), metal rods through the long bones |
| Medication | Bisphosphonates[7] |
| Prognosis | Depends on the type |
| Frequency | 1 in 15,000–20,000 people[8] |
_of_left_forearm.png.webp)
_of_spine%252C_chest%252C_and_pelvis.png.webp)
_of_right_arm.png.webp)
_of_right_femur.png.webp)
The underlying mechanism is usually a problem with connective tissue due to a lack of, or poorly formed, type I collagen.[5]: 1513 In more than 90% of cases, OI occurs due to mutations in the COL1A1 or COL1A2 genes.[14] These mutations may be inherited from a person's parents in an autosomal dominant manner but may also occur spontaneously (de novo).[9][15] There are four clinically defined types: type I, the least severe; type IV, moderately severe; type III, severe and progressively deforming; and type II, perinatally lethal.[9] As of September 2021, 19 different genes are known to cause the 21 documented genetically defined types of OI, many of which are extremely rare and have only been documented in a few individuals.[16][17] Diagnosis is often based on symptoms and may be confirmed by collagen biopsy or DNA sequencing.[10]
Although there is no cure,[10] most cases of OI do not have a major effect on life expectancy,[1]: 461 [15] death during childhood from it is rare,[10] and many adults with OI can achieve a significant degree of autonomy despite disability.[18] Maintaining a healthy lifestyle by exercising, eating a balanced diet sufficient in vitamin D and calcium, and avoiding smoking can help prevent fractures.[19] Genetic counseling may be sought by those with OI to prevent their children from inheriting the disorder from them.[1]: 101 Treatment may include acute care of broken bones, pain medication, physical therapy, mobility aids such as leg braces and wheelchairs,[10] vitamin D supplementation, and, especially in childhood, rodding surgery.[20] Rodding is an implantation of metal intramedullary rods along the long bones (such as the femur) in an attempt to strengthen them.[10] Medical research also supports the use of medications of the bisphosphonate class, such as pamidronate, to increase bone density.[21] Bisphosphonates are especially effective in children,[22] however it is unclear if they either increase quality of life or decrease the rate of fracture incidence.[7]
OI affects only about one in 15,000 to 20,000 people, making it a rare genetic disease.[8] Outcomes depend on the genetic cause of the disorder (its type). Type I (the least severe) is the most common, with other types comprising a minority of cases.[15][23][24] Moderate-to-severe OI primarily affects mobility; if rodding surgery is performed during childhood, some of those with more severe types of OI may gain the ability to walk.[25] The condition has been described since ancient history.[26] The Latinate term osteogenesis imperfecta was coined by Dutch anatomist Willem Vrolik in 1849; translated literally, it means "imperfect bone formation".[26][27]: 683
Signs and symptoms
Orthopedic
The main symptom of osteogenesis imperfecta is fragile, low mineral density bones; all types of OI have some bone involvement.[5] In moderate and especially severe OI, the long bones may be bowed, sometimes extremely so.[28] The weakness of the bones causes them to fracture easily; a study in Pakistan found an average of 5.8 fractures per year in untreated children.[29] Fractures typically occur much less after puberty, but begin to increase again in women after menopause and in men between the ages of 60 and 80.[1]: 486
Joint hypermobility is also a common sign of OI, thought to be because the affected genes are the same as those that cause some types of Ehlers–Danlos syndrome.[5]: 1513 [note 1][30][31]
Otologic
By the age of 50, about 50% of adults with OI experience significant hearing loss, much earlier as compared to the general population.[32] Hearing loss in OI may or may not be associated with visible deformities of the ossicles and inner ear.[33] Hearing loss frequently begins during the second, third, and fourth decades of life, and may be conductive, sensorineural, or a combination of both ("mixed").[34] If hearing loss does not occur by age 50, it is significantly less likely to occur in the years afterwards.[32] Mixed hearing loss is most common among those with OI of all age groups, while conductive hearing loss is most likely to affect older people, with sensorineural hearing loss most likely to affect children.[35]
Although relatively rare, OI-related hearing loss can also begin in childhood; in a study of forty-five children aged four to sixteen, two were found to be affected, aged 11 and 15.[36] In a different 2008 study, the hearing of 41 people with OI was checked. The results showed that 88% of those over 20 years of age had some form of hearing loss, while only 38% of those under 20 did.[37]
Hearing loss is most common in type I OI; it is less common in types III and IV.[1]: 294–296 Other parts of the inner ear may also be affected by OI. causing balance issues; however, only small studies have found links between vertigo and OI.[1]: 308 OI may worsen the outcome of medical treatments which correct hearing loss.[35]
Besides OI's association with sensorineural hearing loss, OI is associated with a number of neurological abnormalities, usually involving the central nervous system, due to deformities in the skeletal structures surrounding it. Neurological complications, especially basilar invagination, may adversely affect life expectancy. In OI, this is most often due to upwards migration of the dens,[38][13]: 106–107 a feature of the C2 vertebra. Neurosurgery may be needed to correct severe abnormalities when they risk the patient's life or cause either great suffering or intolerable neurological deficits.[38][13]: 106–107
Systemic
As its biological causes have been more precisely determined, it has become more widely recognized that while the primary disease process of OI happens in the bones, the most common types of OI—those caused by type I collagen gene mutations—affect virtually all of the human body's organs in some way.[15]
Type I collagen is present throughout the circulatory and respiratory systems: from the ventricles of the heart itself, to the heart valves, to the vasculature,[1]: 329 it is an integral part of the connective tissue of the lungs.[1]: 336 As such, cardiovascular complications, among them aortic insufficiency, aortic aneurysm, and arterial dissections, are sometimes comorbid with OI,[1]: 333 but not as frequently as they are comorbid with Marfan syndrome.[1]: 332
Respiratory illnesses are a major cause of death in OI.[12][1]: 335 The most obvious source of respiratory problems in OI is pulmonary insufficiency caused by problems in the architecture of the thoracic wall.[1]: 341 However, respiratory tract infections, such as pneumonia, are also more fatal among those with OI than the general population.[12][39] Those with more severe ribcage deformities were found to have worse lung restriction in a small-scale 2012 study involving 22 Italian patients with OI types III and IV, plus 26 non-affected controls.[12]
OI—especially its severe form type III—also has effects on the gastrointestinal system. It was found to be associated with recurrent abdominal pain and chronic constipation in two studies on patients affected by OI.[40][41] Chronic constipation is especially common,[1]: 377 and is thought to be aggravated by an asymmetric pelvis (acetabular protrusion).[1]: 377 [41] Especially in childhood, OI-associated constipation may cause a feeling of fullness and associated food refusal, leading to malnutrition.[1]: 377
Classification
There are two typing systems for OI in modern use. The first, created by David Sillence in 1979, classifies patients into four types, or syndromes, according to their clinical presentation, without taking into account the genetic cause of their disease.[42]: 114–115 [43] The second system expands on the Sillence model, but assigns new numbered types genetically as they are found.[44][43] Therefore, people with OI can be described as having both a clinical type and a genetic type, which may or may not be equivalent.[43]
Type I is the most common, and 90% of cases result from mutations to either COL1A1 or COL1A2.[9] Symptoms vary a lot between types, as well as vary from person to person, even in the same family.[45]
As of 2021, 21 types of OI have been defined:[16][17]
| Sillence's four types | ||||||
|---|---|---|---|---|---|---|
| Type | Description | Gene | OMIM | Mode of inheritance | Incidence | Defined[note 2] |
| I | mild | Null COL1A1 allele | 166200 | autosomal dominant, 34% de novo[6] | 1 in 30,000[46] | 1979[43] |
| II | lethal in the perinatal period[5]: 1511 | COL1A1, COL1A2 | 166210 | autosomal dominant, ≈100% de novo[6] | 1 in 40,000[23] to 1 in 100,000[46] | |
| III | severe, progressive and deforming | COL1A1, COL1A2 | 259420 | autosomal dominant, 85% de novo[6] | 1 in 60,000[46] | |
| IV | variable and deforming, but usually with normal sclerae[1]: 294–296 [47] | COL1A1, COL1A2 | 166220 | autosomal dominant, 50% de novo[6] | 1 in 30,000[1]: 21 | |
| Genetically defined types | ||||
|---|---|---|---|---|
| Type | Gene | OMIM | Mode of inheritance | Defined[note 2] |
| V | IFITM5 | 610967 | autosomal dominant[48][49] | 2000[50][note 3] |
| VI | SERPINF1 | 613982 | autosomal recessive[48] | 2002 |
| VII | CRTAP | 610682 | autosomal recessive[48] | 2006 |
| VIII | LEPRE1 | 610915 | autosomal recessive | 2007 |
| IX | PPIB | 259440 | autosomal recessive | 2009 |
| X | SERPINH1 | 613848 | autosomal recessive | 2010 |
| XI | FKBP10 | 610968 | autosomal recessive | 2010 |
| XII | SP7 | 613849 | autosomal recessive | 2010 |
| XIII | BMP1 | 614856 | autosomal recessive | 2012 |
| XIV | TMEM38B | 615066 | autosomal recessive | 2012 |
| XV | WNT1 | 615220 | autosomal recessive | 2013 |
| XVI | CREB3L1 | 616229 | autosomal recessive | 2013 |
| XVII | SPARC | 616507 | autosomal recessive | 2015 |
| XVIII | TENT5A | 617952 | autosomal recessive | 2018 |
| XIX | MBTPS2 | 301014 | X-linked recessive | 2016 |
| XX | MESD | 618644 | autosomal recessive | 2019 |
| XXI | KDELR2 | 619131 | autosomal recessive | 2020 |
Sillence types




Sillence's four types have both a clinical and a genetic meaning; the descriptions below are clinical and can be applied to several genetic types of OI. When used to refer to a genetic as well as a clinical type, it indicates that the clinical symptoms are indeed caused by mutations in the COL1A1 or COL1A2 genes which are inherited in an autosomal dominant fashion.[16]
Type I
Collagen is of normal quality but is produced in insufficient quantities.[5]: 1516 Bones fracture more easily than in the general public, but not as easily as more severe types of OI; there might be scoliosis, albeit mild compared to OI types III and IV, with a lower Cobb angle; the joints may be loose; blue sclerae may be apparent; hearing loss is likely to occur;[47]: Table 1 and there might be a slight decrease in height. Because cases exist missing one or more of these symptoms, OI type I in some cases goes undetected into adulthood.[5]: 1513–1514
Some further split type I into types I–A and I–B, defined as being distinguished by the absence (I–A) or presence (I–B) of dentinogenesis imperfecta (opalescent teeth).[47][55]: 217 People with type I generally have a normal lifespan.[56]
Type II
Collagen is fatally defective at its C-terminus.[5]: 1512 Most cases result in death shortly after birth, or within the first year of life, due to respiratory failure. Another common cause of death is intracranial bleeds from skull fractures present at, or sustained during or shortly after, birth.[5]: 1511 In many cases, the newborn already has multiple broken bones at the time of birth. Type II infants also exhibit severe respiratory problems, and have severely deformed bones. Sixty percent of infants die less than 24 hours after being born, and survival after the first year is extremely unlikely and normally requires mechanical ventilation.[57] In the rare cases of infants who survive their first year of life, severe developmental and motor delays are seen; neither of two infants studied in 2019, both aged around two years, had achieved head control, and both required a ventilator to breathe.[58]
Type II is also known as the "lethal perinatal" form of OI,[59] and is not compatible with survival into adulthood.[57] Due to similarly severely deformed bones, sometimes infants with severe type III are wrongly initially classified as type II; once long-term survival is shown, they are considered as having type III instead.[5]: 1511 [60]
Type III
Collagen quantity is sufficient, but is not of a high enough quality.[5]: 1512 Clinical differentiation between types III and IV is not always simple, and is further confounded by the fact that an untreated adult with type IV may have worse symptoms than a treated adult with type III;[5]: 1511 [61] features only found in type III are its progressively deforming nature[5]: 1511–1512 and the presence of a face with a "triangular" appearance.[62] Another differentiating factor between type III and IV is blue sclerae; in type III, infants commonly have blue sclerae that gradually turn white with age, but blue sclerae are not commonly seen in type IV,[1]: 294–296 although they are seen in 10% of cases.[63]
OI type III causes osteopenic bones that fracture very easily, sometimes even in utero, often leading to hundreds of fractures during a lifetime;[24] early scoliosis that progresses until puberty; dwarfism (a final adult height frequently less than 4 feet or 120 centimetres); loose joints; and possible respiratory problems due to low rib cage volume causing low lung volumes.[5]: 1512
Due to the severity of the issues with the bones, neurological and seizure disorders are more likely to develop in type III.[5]: 1512 Basilar invagination, which puts pressure on the brainstem, may cause or contribute to early death; surgical treatment of it is more complex in OI cases.[5]: 1512 [13]: 106–107
Type IV
Collagen quantity is sufficient, but is not of a high enough quality.[5]: 1512 Type IV is for cases of variable severity, which do not fit into either types III or I.[47] While one of Sillence's required characteristics for type IV was having normal sclerae,[1]: 294–296 [42]: 114 modern classification allows even those with blue sclerae to fit the criteria for type IV if they meet the other clinical requirements of the type.[63]
In type IV, bone deformity can be mild to severe, bones fracture easily (especially before puberty), dwarfism is common, vertebral collapse and scoliosis are evident, and hearing loss is possible,[10] although uncommon.[47][46] Type IV OI is mostly defined in contrast to type III and type I, being the clinical classification for patients somewhere in the middle ground between the two.[5]: 1511 As such, type IV OI is often termed "variable" OI,[42]: 111 with the severity of even those in the same family (so, with the same genetic mutation) differing.[47]
Prepubertal bone fracture rates are another way of clinically assessing type IV OI—those with it tend to have fracture rates of ≈1 per year, compared to ≈3 per year for severe OI (type III).[47]
As in type I, some further split type IV into types IV–A and IV–B, defined again by the absence (IV–A) or presence (IV–B) of dentinogenesis imperfecta.[47][55]: 217
Genetically defined types (types V–XXI)
As of 2020, fifteen types of OI are defined genetically:[16]
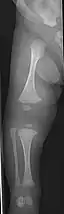
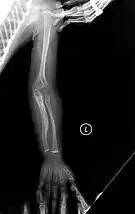
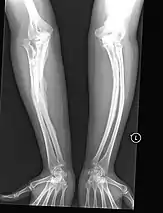
- Type V – Having the same clinical features as type IV, it can be clinically distinguished by observing a "mesh-like" appearance to a bone biopsy under a microscope. Type V can be further distinguished from other types of OI by the "V triad": an opaque band (visible on X-ray) adjacent to the growth plates; hypertrophic calluses (abnormally large masses of bony repair tissue) which form at fracture sites during the healing process; and calcification of the interosseous membrane of the forearm,[50] which may make it difficult to turn the wrist.[1]: 429 Other features of this condition may include pulled elbow, and, as in other types of OI, long bone bowing and hearing loss.[64] Cases of this type are caused by mutations in the IFITM5 gene on chromosome 11p15.5.[64][49] The separation of type V from type IV OI, its clinical type, was initially suggested even before its genetic cause was known, by Glorieux et al. in 2000.[50][65] Type V is relatively common compared to other genetically defined types of OI—4% of OI patients at the genetics department of the Brazilian Hospital de Clínicas de Porto Alegre were found to have it.[66]
- Type VI – With the same clinical features as type III, it is distinguished by bones which have an appearance similar to that seen in osteomalacia.[1]: 168 Type VI is caused by a loss-of-function mutation in the SERPINF1 gene on chromosome 17p13.3.[1]: 170
- Type VII – OI caused by a mutation in the gene CRTAP on chromosome 3p22.3; clinically similar to OI types II and III, depending on affected individual. Type VII was the first recessive OI type confirmed, initially found among First Nations people in Quebec.[67][68]
- Type VIII – OI caused by a mutation in the gene LEPRE1 on chromosome 1p34.2; clinically similar to OI types II and III, depending on affected individual.[69]
- Type IX – OI caused by homozygous or compound heterozygous mutation in the PPIB gene on chromosome 15q22.31.[70]
- Type X – OI caused by homozygous mutation in the SERPINH1 gene on chromosome 11q13.[71]
- Type XI – OI caused by mutations in FKBP10 on chromosome 17q21. The mutations cause a decrease in secretion of trimeric procollagen molecules. Other mutations in this gene can cause autosomal recessive Bruck syndrome, which is similar to OI.[72]
- Type XII – OI caused by a frameshift mutation in SP7 on chromosome 12q13.13. This mutation causes bone deformities, fractures, and delayed tooth eruption.[73]
- Type XIII – OI caused by a mutation in the bone morphogenetic protein 1 (BMP1) gene on chromosome 8p21.3.[74] This mutation causes recurrent fractures, high bone mass, and hypermobile joints.[75]
- Type XIV – OI caused by mutations in the TMEM38B gene on chromosome 9q31.2. This mutation causes recurrent fractures and osteopenia, although the disease trajectory is highly variable.[76]
- Type XV – OI caused by homozygous or compound heterozygous mutations in the WNT1 gene on chromosome 12q13.12. It is autosomal recessive.[75]
- Type XVI – OI caused by mutations in the CREB3L1 gene on chromosome 11p11.2. The homozygous mutation causes prenatal onset of recurrent fractures of the ribs and long bones, demineralization, decreased ossification of the skull, and blue sclerae; it is clinically type II or type III.[77] Family members who are heterozygous for OI XVI may have recurrent fractures, osteopenia and blue sclerae.[77][78]
- Type XVII – OI caused by homozygous mutation in the SPARC gene on chromosome 5q33, causing a defect in the protein osteonectin, which leads to severe disease characterized by generalized platyspondyly, dependence on a wheelchair, and recurrent fractures.[79]
- Type XVIII – OI caused by homozygous mutation in the FAM46A gene on chromosome 6q14.1. Characterized by congenital bowing of the long bones, Wormian bones, blue sclerae, vertebral collapse, and multiple fractures in the first years of life.[80]
- Type XIX – OI caused by hemizygous mutation in the MBTPS2 gene on chromosome Xp22.12. Thus far, OI type XIX is the only known type of OI with an X-linked recessive pattern of inheritance, making it the only type that is more common in males than females. OI type XIX disrupts regulated intramembrane proteolysis, which is critical for healthy bone formation.[81]
- Type XX – OI caused by homozygous mutation in the MESD gene on chromosome 15q25.1. Initial studies of type XX indicate that it may cause global developmental delay, a first among OI types. OI type XX disrupts the Wnt signaling pathway, which is thought to have a role in bone development.[82]
- Type XXI – OI caused by homozygous mutation in the KDELR2 gene on chromosome 7p22.1. Causes disease clinically similar to types II and III, thought to be related to inability of chaperone protein HP47 to unbind from collagen type I, as to do so it needs to bind to the missing ER lumen protein retaining receptor 2 protein encoded by KDELR2.[17]
Given the rapid rate of type discovery, it is extremely likely that there are other genes associated with OI that have yet to be reported.[83]: 491–492
Genetics

Osteogenesis imperfecta is a group of genetic disorders, all of which cause bone fragility. OI has high genetic heterogeneity, that is, many different genetic mutations lead to the same or similar sets of observable symptoms (phenotypes).[84]
The main causes for developing the disorder are a result of mutations in the COL1A1 and/or COL1A2 genes which are jointly responsible for the production of collagen type I.[85] Approximately 90% of people with OI are heterozygous for mutations in either the COL1A1 or COL1A2 genes.[86] There are several biological factors that are results of the dominant form of OI. These factors include: intracellular stress; abnormal tissue mineralization; abnormal cell to cell interactions; abnormal cell-matrix interactions; a compromised cell matrix structure; and, abnormal interaction between non-collagenous proteins and collagen.[87]
Previous research lead to the belief that OI was an autosomal dominant disorder with few other variations in genomes.[8] However, with the lowering of the cost of DNA sequencing in the wake of 2003's Human Genome Project, autosomal recessive forms of the disorder have been identified.[88] Recessive forms of OI relate heavily to defects in the collagen chaperones responsible for production of procollagen and the assembly of the related proteins.[89] Examples of collagen chaperones that are defective in patients with recessive forms of OI include chaperone HSP47 (Cole-Carpenter syndrome) and FKBP65.[44] Mutations in these chaperones result in an improper folding pattern in the collagen 1 proteins which causes the recessive form of the disorder.[44] There are three significant types of OI that are a result of mutations in the collagen prolyl 3-hydroxylation complex (components CRTAP, P3H1, and CyPB).[44] These components are responsible for the modification of collagen α1(l)Pro986.[44] Mutations in other genes such as SP7, SERPINF1, TMEM38B and BMP1 can also lead to irregularly formed proteins and enzymes that result in other recessive types of osteogenesis imperfecta.[44]
Defects in the proteins pigment epithelium-derived factor (PEDF) and bone-restricted interferon-induced transmembrane protein (BRIL) are the causes of type V and VI osteogenesis imperfecta.[90] Defects in these proteins lead to defective bone mineralization which causes the characteristic brittle bones of osteogenesis imperfecta.[90] A single point mutation in the 5′ untranslated region (5′ UTR) of the IFITM5 gene, which encodes BRIL, is linked directly to OI type V.[64][91]
In the rare case of type XIX, first discovered in 2016, OI is inherited as an X-linked genetic disorder, with its detrimental effects resulting ultimately from a mutation in the gene MBTPS2.[81] Genetic research is ongoing, and it is uncertain when all the genetic causes of OI will be identified, as the number of genes that need to be tested to rule out the disorder continue to increase.[83]: 491–492
In a study of 37 families, a 1.3% chance was found that OI recurs in multiple siblings born to two unaffected parents—this is a much higher rate than would be expected if all such recurrences were de novo.[92] The cause is genetic mosaicism; that is, some of, or most of, the germ cells of one parent have a dominant form of OI, but not enough of their somatic cells do to cause symptoms or obvious disability in the parent—the parent's different cells have two (or more) sets of slightly different DNA.[92][5]: 1513 It has been clinically observed that ≈5–10% of cases of OI types II and III are attributable to genetic mosaicism.[1]: 532
Pathophysiology
People with OI are either born with defective connective tissue, born without the ability to make it in sufficient quantities, or, in the rarest genetic types, born with deficiencies in other aspects of bone formation such as chaperone proteins, the Wnt signaling pathway, the BRIL protein, et cetera.[16] In type I the collagen's structure itself is normal, it is just its quantity that is low.[5]: 1516 Types II, III and IV are usually, but not always, related to a deficiency of type I collagen.[93] One possible deficiency arises from an amino acid substitution of glycine to a bulkier amino acid, such as alanine, in the collagen protein's triple helix structure. The larger amino acid side-chains lead to steric effects that creates a bulge in the collagen complex, which in turn influences both the molecular nanomechanics and the interaction between molecules, which are both compromised.[94][95] Depending on both the location of the substitution and the amino acid being used instead, different effects are seen which account for the type diversity in OI despite the same two collagen genes being responsible for most cases.[96][95] Replacements of glycine with serine or cysteine are seen less often in fatal type II OI, while replacements with valine, aspartic acid, glutamic acid, or arginine are seen more often.[95]
At a larger scale, the relationship between the collagen fibrils and hydroxyapatite crystals to form bone is altered, causing brittleness.[96] Bone fractures occur because the stress state within collagen fibrils is altered at the locations of mutations, where locally larger shear forces lead to rapid failure of fibrils even at moderate loads because the homogeneous stress state normally found in healthy collagen fibrils is lost.[94] OI is therefore a multi-scale phenomenon, where defects at the smallest levels of tissues (genetic, nano, micro) domino to affect the macro level of tissues.[94]
Diagnosis
Diagnosis is typically based on medical imaging, including plain X-rays, and symptoms. In severe OI, signs on medical imaging include abnormalities in all extremities and in the spine.[97] As X-rays are often insensitive to the comparatively smaller bone density loss associated with type I OI, DEXA scans may be needed.[5]: 1514
An OI diagnosis can be confirmed through DNA or collagen protein analysis, but in many cases, the occurrence of bone fractures with little trauma and the presence of other clinical features such as blue sclerae are sufficient for a diagnosis. A skin biopsy can be performed to determine the structure and quantity of type I collagen. While DNA testing can confirm the diagnosis, it cannot absolutely exclude it because not all mutations causing OI are yet known and/or tested for.[83]: 491–492 OI type II is often diagnosed by ultrasound during pregnancy, where already multiple fractures and other characteristic features may be visible. Relative to control, OI cortical bone shows increased porosity, canal diameter, and connectivity in micro-computed tomography.[98] OI can also be detected before birth by using an in vitro genetic testing technique such as amniocentresis.[99]
Genetic testing
In order to determine whether osteogenesis imperfecta is present, genetic sequencing of the most common problematic genes, COL1A1, COL1A2, and IFITM5, may be done;[100] if no mutation is found yet OI is still suspected, the other 10+ genes known to cause OI may be tested.[16] Duplication and deletion testing is also suggested to parents who suspect their child has OI.[100] The presence of frameshift mutations caused by duplications and deletions is generally the cause of increased severity of disease.[100]
Differential diagnosis
An important differential diagnosis of OI is child abuse, as both may present to a clinician with multiple fractures in various stages of healing.[5]: 1514 Differentiating them can be difficult, especially when no other characteristic features of OI are present.[1]: 391 This can become an issue in court; in the United States, several child abuse cases were resolved with a finding that osteogenesis imperfecta was the true cause of a child's fractures, leading to lawsuits seeking redress such as Alice Velasquez, et al. v. United States.[1]: 391 [101]
Other differential diagnoses include rickets and osteomalacia, both caused by malnutrition, as well as rare skeletal syndromes such as Bruck syndrome, hypophosphatasia, geroderma osteodysplasticum, and Ehlers–Danlos syndrome.[5]: 1513 [1]: 253–256 Various forms of osteoporosis, such as iatrogenic osteoporosis, idiopathic juvenile osteoporosis, disuse osteoporosis and exercise-related osteoporosis should also be considered as explanations when OI is suspected.[1]: 255–256
Treatment
There is no cure for osteogenesis imperfecta.[10] Maintaining a healthy lifestyle by exercising and avoiding smoking can help prevent fractures.[102] Treatment may include care of broken bones, pain medication, physical therapy, mobility aids such as braces or wheelchairs, and surgery.[102]
Judging the success or failure of treatment can be difficult in OI patients, as decreased bone fracture rates may just be coincidental. While these rates are often used in medical studies to judge treatment efficacy, a Norwegian study of fifteen people with OI emphasized that they feel doctors should consider the whole patient and not just fracture rates.[103]
Acute bone fracture care
Bone fractures are treated in individuals with osteogenesis imperfecta in much the same way as they are treated in the general population—OI bone heals at the same rate as non-OI bone.[1]: 431 A greater emphasis is placed on using lightweight materials to immobilize the fracture, as in moderate or severe types of OI, using heavy casts, such as hip spica casts, can cause fractures at the bones at the boundaries of the cast, as well as generalized osteopenia.[1]: 431 The lightweight cast or splint is then replaced with a removable orthosis after a few weeks and once evidence of union is seen on X-ray.[1]: 431 In order to prevent a nonunion or malunion, all fractures should be immobilized, even if the fracture seems trivial (microfracture),[1]: 439 as people with OI are at greater risk of nonunion.[1]: 438
Bone infections secondary to fractures are treated as and when they occur with the appropriate antibiotics and antiseptics, as in the general population.[1]: 424
Bisphosphonates
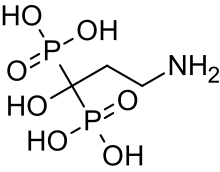
In 1998, an initial observational trial demonstrated the effectiveness of intravenous pamidronate, a bisphosphonate which had previously been used in adults to treat osteoporosis. In severe OI, this trial showed that pamidronate reduced bone pain, prevented new vertebral fractures, reshaped previously fractured vertebral bodies, and reduced the number of long-bone fractures.[104]
Although oral bisphosphonates are more convenient and cheaper, they are not absorbed as well, and intravenous bisphosphonates are generally more effective, although this is under study. Some studies have found oral and intravenous bisphosphonates, such as oral alendronate and intravenous pamidronate, equivalent.[105] In a 2013 double-blind trial of children with mild OI, oral risedronate increased bone mineral densities, and reduced nonvertebral fractures. However, it did not decrease new vertebral fractures.[106][107] A Cochrane review in 2016 concluded that though bisphosphonates seem to improve bone mineral density, it is uncertain whether this leads either to a reduction in bone fractures or improvement in the quality of life of individuals with osteogenesis imperfecta.[7] Even in trials with as many as 125 children, no causal link has been found between bisphosphonates and decreased fracture rates; placebo controlled trials were also unable to prove that they brought about increased strength, motor control or lower pain levels.[87]
Bisphosphonates are not as effective at increasing the bone mineral density of adults.[22]
Nutritional supplements
OI is a genetic disorder and is not caused by insufficient intake of any vitamin or mineral; supplementation cannot cure OI. With that said, people with OI tend to be severely deficient in vitamin D at much higher rates than the general population, and the cause of this is not well understood.[108][109][110] The severity of the deficiency and the likelihood of its occurrence is thought to be related to severity of OI.[109] Vitamin D supplementation may be recommended, at least until levels of 25(OH)D3 in blood return to normal.[108] Below normal levels of vitamin D are also concerning as they may decrease the benefit of bisphosphanates.[108]
Surgery
A surgery of any type inherently carries more risks when done on a patient who has (especially moderate to severe) OI. Skeletal deformities and dentinogenesis imperfecta may hinder access to the airway.[1]: 333 Use of, and weaning off of, mechanical ventilation is also more challenging to carry out on patients with OI.[1]: 333 During the procedure itself or the healing process, defective OI collagen may lead to bleeding diatheses.[1]: 333
The safety of anesthesia is also of more concern among patients with OI,[1]: 333 with anesthetic complications 5.6x more likely to occur when the patient has OI type III.[111] A unique concern of anesthesia in OI is perioperative fracture—fractures sustained due to patient transfer and airway access techniques that, while routine when a patient's bones are strong, may cause injury with brittle OI bones.[112] As an example, due to a 1972 report of a humerus fracture from a sphygmomanometer cuff sustained in an OI patient during surgery, blood pressure monitoring protocols are often modified for patients with OI, with neonatal size cuffs and machine settings being used even in adults;[113]: ¶11.72 further, the least deformed of the patient's limbs is preferred to receive the cuff.[20]: ¶14.23
Rodding

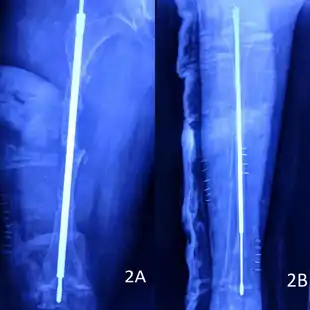
Metal rods can be surgically inserted in the long bones to improve strength, a procedure developed by Harold A. Sofield when he was Chief of Staff at Chicago's Shriners Hospitals for Children, a hospital that offers orthopedic care and surgery to children regardless of their family's ability to pay.[115] Large numbers of children with OI came to Shriners, and Sofield experimented with various methods to strengthen their bones.[116] In 1959, with Edward A. Millar [sic], Sofield wrote a seminal article describing a three-part surgery that seemed radical at the time: precisely breaking the bones ("fragmentation"), putting the resulting bone fragments in a straight line ("realignment"), then placing metal rods into the intramedullary canals of the long bones to stabilize and strengthen them ("rod fixation").[117] His treatment proved useful for increasing the mobility of people with OI, and it has been adopted throughout the world—it became standard surgical treatment for severe OI by 1979, in which year David Sillence found that ≈2⁄3 of the patients he surveyed with OI type III had undergone at least one rodding surgery.[42]: 108
Rodding surgery is often done with the hope that it will offer a path to ambulation, walking, to patients with moderate or severe OI. A 2020 review in The Journal of Bone and Joint Surgery (JB&JS) found it remains broadly popular: ≈2⁄3 of people with OI types III and IV (severe OI) have undergone some form of rodding surgery in their lives, at a mean age of 4+1⁄10 and 7+1⁄2 years respectively;[25]: Table I one possible explanation for a tendency towards earlier intervention in type III is that one half of affected children could not walk at all without the surgery, as their limbs were more bowed, so surgery was sought sooner.[25]
In those with type III OI who had undergone rodding surgery, 79.5% had the femurs and tibias of both legs rodded.[25]: Table I The most common form of rods used are intramedullary (IM) rods, some of which, such as the Fassier–Duval IM rod, are telescoping, meaning that they are designed to grow as the child grows, in an attempt to avoid the necessity of revision surgeries.[118] Telescoping IM rods are widely used,[119] and the common Fassier–Duval IM rod is designed to be used to rod the femur, tibia, and humerus.[120]: 1 The surgery involves breaking the long bones in between one and three (or more)[119]: Figure 4 places, then fixing the rod alongside the bone to keep it straight.[120]: 11
While telescoping IM rods are intended to grow along with both the femur and tibia in developing children; surgeons have a preference to use non-telescoping IM rods, such as Rush rods, in the tibia, which grows less comparatively—the JB&JS review found that while 69.7% of femurs were treated with telescoping IM rods, only 36.9% of tibiae were.[25]: Table IV
While the review in the JB&JS was able to correlate receiving rodding surgery with greater mobility across all types of OI, in patients with type IV, the surgery did not decrease the incidence of broken bones as compared to non-rodded patients—while type IV patients with rodded tibiae experienced 0.93 tibia fractures per year, patients with natural tibiae experienced only 0.81. However, in patients with type III, rodding surgery decreased the average number of tibia fractures per year from 0.84 to 0.57.[25]: Table V
Spinal
Spinal fusion can be performed either as a preventative measure or to correct existing scoliosis, although the inherent fragility of OI bone makes this operation more complex in OI patients than it does with patients who have adolescent idiopathic scoliosis, but normal bone density.[121] Despite the risks, however, three Nemours–duPont orthopedic surgeons who specialize in surgical intervention for osteogenesis imperfecta recommend operating if the curve is greater than 50° after a child is past peak height velocity, as the spine's curve can continue to worsen even into adulthood.[13]: 104
Due to the risk involved, the same surgeons recommend that surgery for basilar impressions and basilar invaginations should only be carried out if the pressure being exerted on the spinal cord and brain stem is causing actual neurological symptoms.[13]: 106–107 Once basilar invagination has become symptomatic, only surgery can halt or reverse the progression of neurological deficits.[1]: 345
Physical therapy
Physical therapy is generally recommended, however individualized protocols are required due to the variability of OI.[1]: 378 Physical therapy is used to strengthen muscles, improve motility, improve flexibility, and help with weight maintenance, although it must be done in a gentle manner to minimize the risk of bone fracture.[1]: 378 In people with OI, exercise often involves water aerobics, light resistance exercises, and walking, if the patient is able.[1]: 378 However, even in patients with mild OI, contact sports, as well as activities likely to put unnecessary stress on the joints, such as jumping, are contraindicated due to the risks they pose.[1]: 378
Individuals with more limited mobility are encouraged to change positions regularly throughout the day; people who sit in a wheelchair most or all of the day are recommended to get out of it every two hours, as a form of exercise, to decrease stiffness, and to prevent pressure ulcers.[1]: 378
Individuals with moderate to severe OI, who require assistive mobility devices and adapted vehicles, face significant barriers to access wheelchair-accessible pools or gyms—they either may not have any in their area, nor the means to get there.[1]: 378 Obesity may be more likely to present among those with severe OI, (especially after the age of 20,) and can, in some, cause further declines in mobility.[122][1]: 371, 373
Tilt table whole body vibration may also be done to increase the mobility of long-term immobilized (bedridden) patients with OI; in at least two cases it helped bedridden children to be able to sit upright.[123][87]
Teeth
More than 1 in 2 people with OI also have dentinogenesis imperfecta (DI)—a congenital abnormality in the formation of dentin, one of the four major components of the human tooth.[124] Dental treatment may pose as a challenge as a result of the various deformities, skeletal and dental, due to OI. Children with OI should go for a dental check-up as soon as their teeth erupt; this may minimize tooth structure loss as a result of abnormal dentine, and they should be monitored regularly to preserve their teeth and oral health.[124]
Many people with OI are treated with bisphosphonates, and there are several possible related complications with dental procedures, for example, medication-related osteonecrosis of the jaw (MRONJ). However, no report of bisphosphonate-related MRONJ in either a child or adult with OI was found in a 2016 Cochrane review of the safety and efficacy of bisphosphonates for OI.[7]
Monoclonal antibodies
Monoclonal antibodies have long been considered for OI, but as of 2021, such therapy has not been approved for OI, neither in the European Union nor in the United States. Thus, it is unclear whether they are safe or effective. Among the monoclonal antibodies that have been studied are romosozumab (targets sclerostin, by Amgen), fresolimumab (TGF-β, Sanofi), blosozumab (sclerostin, Lilly), and setrusumab (sclerostin, begun by Novartis).[125]
Setrusumab, formerly known as BPS-804, is a monoclonal antibody that targets sclerostin, and has been studied in OI specifically more than any of the others. In the body, sclerostin binds to the LRP5 and LRP6 receptors, resulting in inhibition of the Wnt signaling pathway. This decreases bone formation, and is not a problem when a person has healthy bones.[126] It is thought, though, that decreasing the concentration of sclerostin in the body may lead to the formation of more bone, and that is the premise as to why monoclonal antibodies that reduce the concentrations of naturally occurring sclerostin may help strengthen OI bone.[127] While setrusumab was first developed at the pharmaceutical company Novartis, Novartis sold its rights to patent the drug to Mereo Biopharma in 2015, who has continued its development in conjunction with Ultragenyx.[125][128] In 2019, Mereo announced that it had concluded collecting data for its phase II-B trial of setrusumab; the study was completed on 12 November 2020.[125][129] Despite the trial data failing to show improvements in bone density on QCT scans, its primary goal, there were improvements on DXA scans.[130] In a September 2020 press release, Mereo said it was seeking to do a phase III trial in 2021, and had received a Rare Pediatric Disease (RPD) designation from the US Food and Drug Administration (FDA).[131]
Romosozumab, which also is a monoclonal antibody targeting sclerostin, is an approved drug in the US and EU for the treatment of osteoporosis. The pharmaceutical industry analyst Evercore has remarked that "it could wipe out setrusumab's economics", as romosozumab is priced more cheaply than a drug for a rare disease would be, claiming that it will be "vital" to Ultragenyx's profit margins to prove its setrusumab is more efficacious than romosozumab for OI.[125] A clinical trial evaluating romosozumab's efficacy in OI began in September 2020 and as of September 2021 is ongoing.[132] Ultragenyx predicts that its phase 2/3 trials for setrusumab will be completed in 2026.[133][134]
Prevention
As a genetic disorder, the mainstay of twenty-first century prevention of osteogenesis imperfecta is based on preventing affected individuals from being born in the first place. Genetic counseling can help patients and their families determine what types of screening, if any, are right for their situation. Patients can consider preimplantation genetic diagnosis after in vitro fertilization to select fertilized embryos which are not affected.[1]: 247–248 Common mutations which cause OI may be caught by exome sequencing and whole genome sequencing. If a pregnancy is already in progress, the procedure of amniocentesis may be undergone to see if the fetus is affected.[1]: 247 If affected, it is up to the family to consider whether or not they want to terminate the pregnancy and try again—raising questions of medical ethics and a woman's right to choose.[135][136]
Without intervention, patients with the most common mutations causing osteogenesis imperfecta have a 50% chance per gestation of passing on the disorder, as these mutations are inherited in an autosomal dominant pattern of Mendelian inheritance.[1]: 247 Those with the rare autosomal recessive forms of OI have a 25% chance of passing on the disorder. Genetic testing of the affected members of the family can be used to determine which inheritance pattern applies.[1]: 101
As OI type I may be difficult to detect in a newborn child, the cord blood of the child can be tested to determine if it has been passed on if the family has already rejected the more invasive genetic screening methods.[1]: 247 In more severe cases, the diagnosis may be able to be done via ultrasound, especially if OI is already a possibility.[1]: 248 An ethical concern with prenatal screening for OI often arises when parents inquire as to how severely affected their child will be—such questions are as yet difficult to answer conclusively.[1]: 382
If a non-affected person has already had a child with OI, there is a greater likelihood (although still quite remote), that their future children will have OI due to genetic mosaicism.[92][1]: 100, 1513
The disability rights critique of prenatal screening for OI, held by some bioethicists and some affected individuals, negatively compares it to eugenics, with even those not opposed to abortion opposing selective abortions on the ethical ground that their existence betrays the belief that the lives of those with OI are "less worth living [and] less valuable".[1]: 388
Prognosis
The prognosis of osteogenesis imperfecta depends entirely on its type (see § Classification).
Life expectancy
In the mild form of the disorder, type I, the life expectancy of patients is near that of the general population.[1]: 461 In type II, however, patients only very rarely live past the age of two, and typically die in their first weeks of life.[5]: 1511 Assessment of the life expectancy of patients with types III and IV is more complicated, as lifestyle choices can cause fatally traumatic injuries that would not have otherwise occurred, or not been fatal in the general population. Life expectancy in type IV OI is thought to be close to normal, but in type III it is lower than in the general population.[48]
A 2016 study of data in Denmark's National Patient Register found that across all types of OI, all-cause mortality was three times higher, leading to a loss of around seven years in females and nine years in males.[15] A 1996 study published in the British Medical Journal found that mortality in type III OI is significantly higher, with many patients dying in their 20's, 30's, and 40's; patients who survive to the age of 10 were further found to have longer life expectancy than newborns.[137]
Mobility
People with mild (type I) OI as adults need few pieces of adaptive equipment, although in infancy they reach motor milestones at a significant delay compared to the general population.[1]: 477
With adaptive equipment such as crutches, motorized wheelchairs, splints, reach extenders, and/or modifications to the home, many individuals with moderate to severe OI can achieve or maintain a significant degree of independence.[18][1]: 488 With treatment and physical therapy, the maximum levels of mobility are expected to be unassisted community walking for type I, household or exercise walking for type III, and household or community walking for type IV; due to the variability of OI between individuals, mobility achieved varies and may be below this expected maximum.[1]: 476
Epidemiology
In the United States, the incidence of osteogenesis imperfecta is estimated to be one per 20,000 live births.[138] An estimated 20,000 to 50,000 people are affected by OI in the United States.[139]
The most common types are I, II, III, and IV, while the rest are very rare.[140] Type I is the most common and has been reported to be around three times more common than type II. The prevalence of types III and IV is less certain.[23] In a 1989 study in Denmark, type I was found to comprise 71% of cases and type II 12% of cases, with other types comprising the other 17%.[15] In a 2015 study in Sweden, type I was nearly six times more common than type III and nearly four times more common than type IV.[24]
Most people with OI receive it from a parent, but in many cases, it is a brand new (de novo or "sporadic") mutation in a family. Among a study of patients with survivable types of OI, OI type III is most often de novo (85%), followed by type IV (50%) and type I (34%).[6]: Table 1
Some populations can have a higher incidence of OI than would be otherwise expected if they have a larger than average number of carriers of the recessive forms of the disease.[1]: 20–21 [141]
History


.jpg.webp)
The condition, or types of it, has had various other names over the years and in different nations; "osteogenesis imperfecta" has, however, been the most widely accepted name for the condition since the late 20th century. Among some of the most common alternatives are "fragilitas ossium";[2] "Ekman–Lobstein syndrome", and "Vrolik syndrome", both eponyms; and, the colloquialism, "brittle bone disease".[1]
Earliest recorded cases
OI has been identified in an ancient Egyptian infant mummified in around 1000 BC, originally dismissed by archaeologists as containing the remains of a monkey.[142][143]: 161 The Norse king Ivar the Boneless, who lived c. 800 CE,[144] is speculated to have had OI as well.[145]
Nicolas de Malebranche is oft credited as being the first person to describe the physical characteristics of OI in his 1688 book The Search after Truth, in which he describes a man who has had his "bones broken in the places a murderer's would be" all his life. His confident description of the pathology of the disorder, however, which creates what he termed «enfants monstrueux» ("monstrous children"), is scientifically void—he wrote that it was due to the mother's antepartum viewership of a public execution by breaking wheel.[27]: 683 [143]: 165–168 [146]
The earliest modern scientific studies of OI began in 1788 by Olof Jakob Ekman, who described the condition, which he termed "osteomalacia congenital", in his doctoral thesis and mentioned cases of it going back to 1678, all in the same family, through three generations.[147] Ekman's description of the condition mentioned dwarfism, bone fragility, and bowing of the long bones.[148]: 763 In 1831, Edmund Axmann gave a detailed description of it in himself and his two brothers, being the first to mention blue sclerae as a characteristic sign of OI.[27]: 683 Jean Lobstein first described the mild form of the condition, today known as type I, in 1833, calling it "osteopsathyrosis idiopathica".[3]: 347
It was not until 1912 that hearing loss was positively recognized as a symptom of OI, first mentioned in a brief paper by the English physician Charles Allen Adair-Dighton.[149][143]: 168–169
Of the term
.jpg.webp)
Willem Vrolik, a Dutch anatomist who was also curator of the "Museum Vrolikianum", which made him privy to many specimens of bodies having birth defects, coined the term "osteogenesis imperfecta"[27]: 683 in his bilingual Latin and Dutch language book on teratology, Illustrations of Human and Mammalian Embryogenesis, first published in 1849.[150]
Included is a description of the remains of an infant who had what is now known as perinatally fatal OI type II[3]: 347 [1]: 5 (as verified in a 1998 re-examination of the remains by Baljet et al.).[151] The remains were first given to Vrolik's father, who could not make sense of them. Vrolik described poorly mineralized bones, bowed long bones, and fractures in various states of healing.[152] Vrolik correctly determined that what he termed OI in the infant was not caused by secondary rickets, but a congenital abnomality causing primary osteopenia; he theorized this was due to a lack of "intrinsic generative energy".[151]
Of its classification
Classification of OI has also evolved as scientific understanding of it has improved. Before the advent of modern genetic testing, OI was classified in two broad groups: osteogenesis imperfecta congenita, and osteogenesis imperfecta tarda, a division first proposed by the German physician E. Looser in 1906.[2][153] Congenita was used to describe the modern clinical types II, III, and some cases of IV, where upon birth the condition was obvious, either due to bowing of the limbs or due to fractures sustained in utero.[42] Tarda was used to classify the modern OI type I and some cases of type IV, where the inherent fragility of the bones did not become clear until long after birth.[2] The idea that these "late" and "prenatal" forms were manifestations of the same disorder was first proposed in 1897 by Martin Benno Schmidt;[154] by the 1950s this fact was well accepted.[3]: 346
The modern system of four types (I, II, III, IV), meanwhile, were introduced in a paper by David Sillence, Alison Senn, and David Danks in the Journal of Medical Genetics in 1979,[42][155] and have since become standard terms among doctors, patients, and researchers.[43][47][156] The modern genetic types, (those with numbers greater than IV,) have come into use as more and more recessively inherited forms of OI have been discovered since the discovery of the first one by Roy Morello et al. in 2006.[46][8][67] In 2010, the International Nomenclature Group for Constitutional Disorders of the Skeleton (INCDS) "freed" the Sillence types from molecular reference, acquiescing to their new clinical-first role in the wake of what was to them a "surprising" increase in the number of genetic causes of OI.[156][47]
Writing for the Annual Review of Genetics in 2012, Drs. Peter Byers and Shawna M. Pyott lamented how the expansion of the number of types to include genetic types has created a system that "grew like Topsy".[83]: 492 They suggest that it may indeed be impossible to create a system which is useful for clinicians and which accurately describes the genetic cause of a person's OI, with attempts always prioritizing one use at the expense of the other.[83]: 492
Society and culture
Much medical research has been done into the causes of osteogenesis imperfecta, benefiting not only those with OI but medicine more broadly; in the ten years between 2006 and 2016, the many discoveries of non-collagen related recessive gene mutations, which still led in those who have them to the clinical signs of OI, led to numerous breakthroughs in medical understanding of the process of healthy bone development.[8]
Other animals
In dogs, OI is an autosomal recessive condition, meaning that dogs with two copies of the allele will be affected.[157] Many breed organizations and veterinarians offer OI tests to tell if a dog is a carrier of OI.[157][158] Dogs who are heterozygous for OI should only be bred to non-carriers. Homozygous carriers should never be bred, unless it is to a non-carrier.[158]
Naturally occurring mutations causing OI have been found in Golden Retrievers, Dachshunds, and Beagles. OI has also been identified in zebrafish and mice.[159]
Although dogs, mice, fish, and humans are not genetically identical, some of these animal models have been officially recognized to represent the varying types of OI in humans. For example, homozygous oim/oim mice experience spontaneous bone fractures, small body size, and kyphosis, making them a model of OI type III. Meanwhile, heterozygous oim/+ mice appear normal but have bones which are quite a bit weaker than wild mice, making them a model for OI type I.[159][160] As in human OI, the location on the gene which is mutated affects the severity of resulting disease—the G859C Col1a1 mouse is a model for OI type II as affected mice all die in the perinatal period.[159]
Animal testing on identified animal models may lead to human therapies for OI.[159]
Explanatory notes
- One OI mutation also causes combined Ehler–Danlos syndrome: "OIEDS1". (Cabral, et al. 2005, Rasmussen 2020)
- When year is uncited, OMIM is the source.
- Genetic cause not determined until 2012. (Cho, et al. 2012)
References
- Shapiro JR, Byers PH, Glorieux FH, Sponseller PD (2014). Osteogenesis imperfecta: a translational approach to brittle bone disease (2nd ed.). London: Elsevier Inc. ISBN 978-0-12-397165-4. OCLC 876364090.
- Roger S, Francis MJ, Houghton GR (1983). The brittle bone syndrome: osteogenesis imperfecta. London: Butterworth-Heinemann. p. 4. ISBN 978-0-407-00211-1. OCLC 9784850.
- Davel JG, Fichardt T, Van Der Spuy D (October 1956). "Osteogenesis imperfecta". Archives of Disease in Childhood. 31 (159): 346–353. doi:10.1136/adc.31.159.346. PMC 2011939. PMID 13363481.
-
- "osteogenesis". Lexico UK English Dictionary. Oxford University Press. Archived from the original on 23 August 2021. "osteogenesis imperfecta". Lexico UK English Dictionary UK English Dictionary. Oxford University Press. Archived from the original on 23 August 2021.
- "osteogenesis". Merriam-Webster Dictionary. Retrieved 23 August 2021. "osteogenesis imperfecta". Merriam-Webster Dictionary. Retrieved 23 August 2021.
- Rowe DW (2008). "Osteogenesis imperfecta". Principles of bone biology (3rd ed.). Amsterdam: Elsevier. pp. 1511–1531. ISBN 978-0-12-373884-4. OCLC 267135745. Archived from the original on 20 September 2021. Retrieved 15 August 2021.
Dentinogenesis imperfecta (DI) is most frequent in OI types III and IV, and overall, affects about 15% of OI patients among the different phenotypes.
- Zhytnik L, Maasalu K, Duy BH, Pashenko A, Khmyzov S, Reimann E, et al. (March 2019). "De novo and inherited pathogenic variants in collagen-related osteogenesis imperfecta". Molecular Genetics & Genomic Medicine. 7 (3): e559. doi:10.1002/mgg3.559. PMC 6418448. PMID 30675999.
- Dwan K, Phillipi CA, Steiner RD, Basel D (October 2016). "Bisphosphonate therapy for osteogenesis imperfecta". The Cochrane Database of Systematic Reviews. 2016 (10): CD005088. doi:10.1002/14651858.CD005088.pub4. PMC 6611487. PMID 27760454.
- Forlino A, Marini JC (April 2016). "Osteogenesis imperfecta". Lancet. 387 (10028): 1657–1671. doi:10.1016/S0140-6736(15)00728-X. PMC 7384887. PMID 26542481.
- "Osteogenesis imperfecta". Genetics Home Reference. U.S. National Library of Medicine, National Institutes of Health. 18 August 2020. Archived from the original on 29 August 2021. Retrieved 15 August 2021.
- Lee B, Krakow D (July 2019). "Osteogenesis Imperfecta Overview". National Institute of Arthritis and Musculoskeletal and Skin Diseases. National Institutes of Health. Archived from the original on 9 May 2021. Retrieved 21 August 2021.
- McNeeley MF, Dontchos BN, Laflamme MA, Hubka M, Sadro CT (December 2012). "Aortic dissection in osteogenesis imperfecta: case report and review of the literature". Emergency Radiology. 19 (6): 553–556. doi:10.1007/s10140-012-1044-1. PMID 22527359. S2CID 11109481.
- LoMauro A, Pochintesta S, Romei M, D'Angelo MG, Pedotti A, Turconi AC, Aliverti A (27 April 2012). "Rib cage deformities alter respiratory muscle action and chest wall function in patients with severe osteogenesis imperfecta". PLOS ONE. 7 (4): e35965. Bibcode:2012PLoSO...735965L. doi:10.1371/journal.pone.0035965. PMC 3338769. PMID 22558284.
- Wallace MJ, Kruse RW, Shah SA (February 2017). "The Spine in Patients With Osteogenesis Imperfecta". The Journal of the American Academy of Orthopaedic Surgeons. 25 (2): 100–109. doi:10.5435/JAAOS-D-15-00169. PMID 28009707.
- Shapiro JR, Sponseller PD (23 August 2005). "10—Osteogenesis Imperfecta". In Kleerekoper M, Siris ES, McClung M (eds.). The Bone and Mineral Manual: A Practical Guide. Elsevier Inc. p. 45. ISBN 978-0-08-047074-0. Archived from the original on 20 September 2021. Retrieved 17 September 2021.
- Folkestad L, Hald JD, Canudas-Romo V, Gram J, Hermann AP, Langdahl B, et al. (December 2016). "Mortality and Causes of Death in Patients With Osteogenesis Imperfecta: A Register-Based Nationwide Cohort Study". Journal of Bone and Mineral Research. 31 (12): 2159–2166. doi:10.1002/jbmr.2895. PMID 27345018. S2CID 32196304. Archived from the original on 26 August 2021. Retrieved 26 August 2021.
- Marom R, Rabenhorst BM, Morello R (October 2020). "Osteogenesis imperfecta: an update on clinical features and therapies". European Journal of Endocrinology. 183 (4): R95–R106. doi:10.1530/EJE-20-0299. PMC 7694877. PMID 32621590. Archived from the original on 17 August 2021. Retrieved 17 August 2021.
- van Dijk FS, Semler O, Etich J, Köhler A, Jimenez-Estrada JA, Bravenboer N, et al. (November 2020). "Interaction between KDELR2 and HSP47 as a Key Determinant in Osteogenesis Imperfecta Caused by Bi-allelic Variants in KDELR2". American Journal of Human Genetics. 107 (5): 989–999. doi:10.1016/j.ajhg.2020.09.009. PMC 7675035. PMID 33053334. Archived from the original on 20 September 2021. Retrieved 29 August 2021.
- Donohoe M (2020). "Therapy, Orthotics and Assistive Devices for Osteogenesis Imperfecta". In Kruse RW (ed.). Osteogenesis Imperfecta. Osteogenesis Imperfecta: A Case-Based Guide to Surgical Decision-Making and Care. Springer International Publishing. pp. 21–37. doi:10.1007/978-3-030-42527-2_3. ISBN 978-3-030-42526-5. S2CID 219518604.
- "What People With Osteogenesis Imperfecta Need To Know About Osteoporosis". Osteoporosis and Related Bone Diseases National Resource Center. National Institutes of Health. November 2018. Archived from the original on 18 March 2021. Retrieved 18 September 2021.
- Esposito P, Wallace MJ (2020), Kruse RW (ed.), "Osteogenesis Imperfecta Surgical Management of the Femur and Knee" (ePub), Osteogenesis Imperfecta: A Case-Based Guide to Surgical Decision-Making and Care, Springer International Publishing, pp. 147–182, doi:10.1007/978-3-030-42527-2_10, ISBN 978-3-030-42527-2, S2CID 219515805
- Harrington J, Sochett E, Howard A (December 2014). "Update on the evaluation and treatment of osteogenesis imperfecta". Pediatric Clinics of North America. 61 (6): 1243–1257. doi:10.1016/j.pcl.2014.08.010. PMID 25439022.
- Chevrel G, Schott AM, Fontanges E, Charrin JE, Lina-Granade G, Duboeuf F, et al. (February 2006). "Effects of oral alendronate on BMD in adult patients with osteogenesis imperfecta: a 3-year randomized placebo-controlled trial". Journal of Bone and Mineral Research. 21 (2): 300–306. doi:10.1359/JBMR.051015. PMID 16418786. S2CID 34089615.
- Subramanian S, Viswanathan VK (2021). Osteogenesis Imperfecta. StatPearls Publishing. StatPearls. PMID 30725642. Archived from the original on 21 October 2020. Retrieved 30 July 2019. Last Update: February 3, 2019.
- Lindahl K, Åström E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren Ö, Kindmark A (August 2015). "Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta". European Journal of Human Genetics. 23 (8): 1042–1050. doi:10.1038/ejhg.2015.81. PMC 4795106. PMID 25944380.
- Rodriguez Celin M, Kruger KM, Caudill A, Nagamani SC, Harris GF, Smith PA (11 September 2020). "A Multicenter Study of Intramedullary Rodding in Osteogenesis Imperfecta". JB & JS Open Access. 5 (3): e20.00031. doi:10.2106/JBJS.OA.20.00031. PMC 7489747. PMID 32984750.
- Kelly EB (2012). Encyclopedia of Human Genetics and Disease. ABC-CLIO. p. 613. ISBN 9780313387135. Archived from the original on 5 November 2017.
- Dang Do AN, Marini JC (27 January 2021). "Osteogenesis imperfecta". In Carey JC, Cassidy SB, Battaglia A, Viskochil D (eds.). Cassidy and Allanson's Management of Genetic Syndromes. John Wiley & Sons. pp. 683–705. ISBN 978-1-119-43267-8. Archived from the original on 29 August 2021. Retrieved 29 August 2021.
- Johnson MT, Morrison S, Heeger S, Mooney S, Byers PH, Robin NH (February 2002). "A variant of osteogenesis imperfecta type IV with resolving kyphomelia is caused by a novel COL1A2 mutation". Journal of Medical Genetics. 39 (2): 128–132. doi:10.1136/jmg.39.2.128. PMC 1735034. PMID 11836364.
- Atta I, Iqbal F, Lone SW, et al. "Journal of the College of Physicians and Surgeons Pakistan" (PDF). Journal of the College of Physicians and Surgeons Pakistan. 24 (9): 653–657. Archived (PDF) from the original on 17 August 2021. Retrieved 17 August 2021.
- Cabral WA, Makareeva E, Colige A, Letocha AD, Ty JM, Yeowell HN, et al. (May 2005). "Mutations near amino end of alpha1(I) collagen cause combined osteogenesis imperfecta/Ehlers-Danlos syndrome by interference with N-propeptide processing". The Journal of Biological Chemistry. 280 (19): 19259–19269. doi:10.1074/jbc.m414698200. PMID 15728585. Archived from the original on 20 September 2021. Retrieved 17 August 2021.
- Rasmussen SA (2 December 2020). "Combined osteogenesis imperfecta and Ehlers–Danlos syndrome 1; OIEDS1". Online Mendelian Inheritance in Man. Johns Hopkins University. 619115. Archived from the original on 20 September 2021. Retrieved 17 August 2021.
- Paterson CR, Monk EA, McAllion SJ (April 2001). "How common is hearing impairment in osteogenesis imperfecta?". The Journal of Laryngology and Otology. 115 (4): 280–282. doi:10.1258/0022215011907442. PMID 11276328. S2CID 12303464. Archived from the original on 20 September 2021. Retrieved 15 August 2021.
- Vernick D (2 November 2005). "OI Issues: Hearing Loss". Archived from the original on 20 January 2012. Retrieved 4 November 2018.
- Santos F, McCall AA, Chien W, Merchant S (December 2012). "Otopathology in Osteogenesis Imperfecta". Otology & Neurotology. 33 (9): 1562–1566. doi:10.1097/MAO.0b013e31826bf19b. PMC 3498599. PMID 22996160.
- Pillion JP, Vernick D, Shapiro J (14 December 2011). "Hearing loss in osteogenesis imperfecta: characteristics and treatment considerations". Genetics Research International. Hindawi. 2011: 983942. doi:10.4061/2011/983942. PMC 3335494. PMID 22567374.
- Kuurila K, Grénman R, Johansson R, Kaitila I (July 2000). "Hearing loss in children with osteogenesis imperfecta". European Journal of Pediatrics. 159 (7): 515–519. doi:10.1007/s004310051322. PMID 10923226. S2CID 8729406. Archived from the original on 15 August 2021. Retrieved 15 August 2021.
- Pillion JP, Shapiro J (September 2008). "Audiological findings in osteogenesis imperfecta". Journal of the American Academy of Audiology. 19 (8): 595–601. doi:10.3766/jaaa.19.8.3. PMID 19323351.
- Khandanpour N, Connolly DJ, Raghavan A, Griffiths PD, Hoggard N (1 December 2012). "Craniospinal abnormalities and neurologic complications of osteogenesis imperfecta: imaging overview". Radiographics. 32 (7): 2101–2112. doi:10.1148/rg.327125716. PMID 23150860. Archived from the original on 18 August 2021. Retrieved 18 August 2021.
- Turkalj M, Miranović V, Lulić-Jurjević R, Gjergja Juraški R, Primorac D (25 September 2017). "Cardiorespiratory complications in patients with osteogenesis imperfecta". Paediatria Croatica. 61 (3): 106–111. ISSN 1330-1403. Archived from the original on 20 September 2021. Retrieved 29 August 2021.
- Violas P, Fassier F, Hamdy R, Duhaime M, Glorieux FH (September 2002). "Acetabular protrusion in osteogenesis imperfecta". Journal of Pediatric Orthopedics. 22 (5): 622–625. doi:10.1097/01241398-200209000-00010. PMID 12198464. S2CID 37895736.
- Lee JH, Gamble JG, Moore RE, Rinsky LA (September 1995). "Gastrointestinal problems in patients who have type-III osteogenesis imperfecta". The Journal of Bone and Joint Surgery. American Volume. 77 (9): 1352–1356. doi:10.2106/00004623-199509000-00010. PMID 7673285.
- Sillence DO, Senn A, Danks DM (April 1979). "Genetic heterogeneity in osteogenesis imperfecta". Journal of Medical Genetics. 16 (2): 101–116. doi:10.1136/jmg.16.2.101. PMC 1012733. PMID 458828.
- Van Dijk FS, Pals G, Van Rijn RR, Nikkels PG, Cobben JM (1 January 2010). "Classification of Osteogenesis Imperfecta revisited". European Journal of Medical Genetics. 53 (1): 1–5. doi:10.1016/j.ejmg.2009.10.007. PMID 19878741. Archived from the original on 21 August 2021. Retrieved 21 August 2021.
In 1979 Sillence proposed a classification of Osteogenesis Imperfecta (OI) in OI types I, II, III and IV. In 2004 and 2007 this classification was expanded with OI types V–VIII because of distinct clinical features and/or different causative gene mutations.
- Marini JC, Blissett AR (August 2013). "New genes in bone development: what's new in osteogenesis imperfecta". The Journal of Clinical Endocrinology and Metabolism. 98 (8): 3095–3103. doi:10.1210/jc.2013-1505. PMC 3733862. PMID 23771926.
- McKusick VA, Hamosh A (23 October 2018). "Osteogenesis imperfecta, type I; OI1". Online Mendelian Inheritance in Man. Johns Hopkins University. 166200. Archived from the original on 26 March 2021. Retrieved 17 August 2021.
- van Dijk FS, Cobben JM, Kariminejad A, Maugeri A, Nikkels PG, van Rijn RR, Pals G (December 2011). "Osteogenesis Imperfecta: A Review with Clinical Examples". Molecular Syndromology. 2 (1): 1–20. doi:10.1159/000332228. PMC 3343766. PMID 22570641.
- Van Dijk FS, Sillence DO (June 2014). "Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment". American Journal of Medical Genetics. Part A. 164A (6): 1470–1481. doi:10.1002/ajmg.a.36545. PMC 4314691. PMID 24715559.
- Steiner RD, Pepin MG, Byers PH, Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (6 May 2021). "COL1A1/2 Osteogenesis Imperfecta". In Adam MP, Ardinger HH, Pagon RA, et al. (eds.). GeneReviews [Internet]. University of Washington, Seattle. PMID 20301472. Archived from the original on 18 January 2017. Retrieved 26 August 2021.
- Shapiro JR, Lietman C, Grover M, Lu JT, Nagamani SC, Dawson BC, et al. (July 2013). "Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation". Journal of Bone and Mineral Research. 28 (7): 1523–1530. doi:10.1002/jbmr.1891. PMC 3688672. PMID 23408678.
- Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, et al. (September 2000). "Type V osteogenesis imperfecta: a new form of brittle bone disease". Journal of Bone and Mineral Research. 15 (9): 1650–1658. doi:10.1359/jbmr.2000.15.9.1650. PMID 10976985. S2CID 13748803.
- Rudd A (26 March 2016). "Jordanne Whiley: 'To achieve perfection is impossible. You should be who you are because someone will find you attractive'". The Times. ISSN 0140-0460. Retrieved 26 March 2022.
- Harvey K (6 March 2015). "The Middle star says it's OK to be a nerd". Stuff. New Zealand: Stuff Ltd. Retrieved 25 May 2018.
- "Schauspieler und Kämpfer für Behindertenrechte: Peter Radtke ist tot" [Actor and campaigner for disability rights Peter Radtke has died]. Der Spiegel (in German). 30 November 2020. ISSN 2195-1349. Retrieved 26 March 2022.
- "Theresia Haidlmayr". Behinderte Menschen in Not (in Austrian German). Retrieved 26 March 2022.
- Singer RB, Ogston SA, Paterson CR (2001). "Mortality in various types of osteogenesis imperfecta". Journal of Insurance Medicine. 33 (3): 216–220. PMID 11558400. Archived from the original on 15 August 2021. Retrieved 15 August 2021.
- "Osteogenesis Imperfecta: Types, Symptoms & Management". Cleveland Clinic. 5 May 2021. Archived from the original on 17 August 2021. Retrieved 17 August 2021.
- Basel D, Steiner RD (June 2009). "Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition" (PDF). Genetics in Medicine. 11 (6): 375–385. doi:10.1097/GIM.0b013e3181a1ff7b. PMID 19533842. Archived (PDF) from the original on 20 September 2021. Retrieved 19 August 2021.
- Nadar R, Saraff V, Hogler W, Desai M, Shaw N (31 July 2019). "Determinants of survival in osteogenesis imperfecta (OI) Type II". Bone Abstracts. 7: 31. doi:10.1530/boneabs.7.P31. ISSN 2052-1219. S2CID 208412310. Archived from the original on 20 September 2021. Retrieved 19 August 2021.
- Zisa D, Shabbir A, Mastri M, Suzuki G, Lee T (November 2009). "Intramuscular VEGF repairs the failing heart: role of host-derived growth factors and mobilization of progenitor cells". American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 297 (5): R1503–R1515. doi:10.1152/ajpregu.00227.2009. PMC 2777764. PMID 19759338.
- Ben Amor IM, Glorieux FH, Rauch F (6 September 2011). "Genotype-phenotype correlations in autosomal dominant osteogenesis imperfecta". Journal of Osteoporosis. 2011: 540178. doi:10.4061/2011/540178. PMC 3170785. PMID 21912751.
For example, it can occur that a fetus is diagnosed with OI type II, the lethal form, on prenatal ultrasound, but after birth turns out to be less severely affected than initially thought and is eventually reclassified as OI type III or even type VI, if adequate management is initiated early on.
- Georgescu I, Vlad C, Gavriliu TŞ, Dan S, Pârvan AA (June 2013). "Surgical treatment in Osteogenesis Imperfecta - 10 years experience". Journal of Medicine and Life. 6 (2): 205–213. PMC 3725451. PMID 23904885.
- Chen H (2006). "Triangular face". Atlas of genetic diagnosis and counseling. Totowa, NJ: Humana Press. p. 771. ISBN 978-1-58829-681-8. Archived from the original on 8 June 2013.
- Johnson MT, Morrison S, Heeger S, Mooney S, Byers PH, Robin NH (February 2002). "A variant of osteogenesis imperfecta type IV with resolving kyphomelia is caused by a novel COL1A2 mutation". Journal of Medical Genetics. 39 (2): 128–132. doi:10.1136/jmg.39.2.128. PMC 1735034. PMID 11836364.
The persistent pale blue sclera is uncommon in OI type IV, but can be seen in up to 10% of affected subjects.
- Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, et al. (August 2012). "A single recurrent mutation in the 5'-UTR of IFITM5 causes osteogenesis imperfecta type V". American Journal of Human Genetics. 91 (2): 343–348. doi:10.1016/j.ajhg.2012.06.005. PMC 3415533. PMID 22863190.
- Cao YJ, Wei Z, Zhang H, Zhang ZL (12 June 2019). "Expanding the Clinical Spectrum of Osteogenesis Imperfecta Type V: 13 Additional Patients and Review". Frontiers in Endocrinology. 10: 375. doi:10.3389/fendo.2019.00375. PMC 6581704. PMID 31244780.
- Brizola E, Mattos EP, Ferrari J, Freire PO, Germer R, Llerena JC, Félix TM (October 2015). "Clinical and Molecular Characterization of Osteogenesis Imperfecta Type V". Molecular Syndromology. 6 (4): 164–172. doi:10.1159/000439506. PMC 4662268. PMID 26648832.
- Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, et al. (October 2006). "CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta". Cell. 127 (2): 291–304. doi:10.1016/j.cell.2006.08.039. PMID 17055431. S2CID 8123837.
- Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, et al. (December 2006). "Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta". The New England Journal of Medicine. 355 (26): 2757–2764. doi:10.1056/NEJMoa063804. PMC 7509984. PMID 17192541.
- Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, et al. (March 2007). "Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta". Nature Genetics. 39 (3): 359–365. doi:10.1038/ng1968. PMC 7510175. PMID 17277775.
- van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, et al. (October 2009). "PPIB mutations cause severe osteogenesis imperfecta". American Journal of Human Genetics. 85 (4): 521–527. doi:10.1016/j.ajhg.2009.09.001. PMC 2756556. PMID 19781681.
- Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, et al. (March 2010). "Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta". American Journal of Human Genetics. 86 (3): 389–398. doi:10.1016/j.ajhg.2010.01.034. PMC 2833387. PMID 20188343.
- Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, et al. (April 2010). "Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta". American Journal of Human Genetics. 86 (4): 551–559. doi:10.1016/j.ajhg.2010.02.022. PMC 2850430. PMID 20362275.
- Sam JE, Dharmalingam M (2017). "Osteogenesis Imperfecta". Indian Journal of Endocrinology and Metabolism. 21 (6): 903–908. doi:10.4103/ijem.IJEM_220_17. PMC 5729682. PMID 29285457.
- Shapiro JR (2014). "Clinical and Genetic Classification of Osteogenesis Imperfecta and Epidemiology". Osteogenesis Imperfecta. Elsevier. pp. 15–22. doi:10.1016/b978-0-12-397165-4.00002-2. ISBN 9780123971654.
- Sam JE, Dharmalingam M (1 November 2017). "Osteogenesis Imperfecta". Indian Journal of Endocrinology and Metabolism. 21 (6): 903–908. doi:10.4103/ijem.IJEM_220_17. PMC 5729682. PMID 29285457.
- Shaheen R, Alazami AM, Alshammari MJ, Faqeih E, Alhashmi N, Mousa N, et al. (October 2012). "Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation". Journal of Medical Genetics. 49 (10): 630–635. doi:10.1136/jmedgenet-2012-101142. PMID 23054245. S2CID 35542661.
- Lindahl K, Åström E, Dragomir A, Symoens S, Coucke P, Larsson S, et al. (September 2018). "Homozygosity for CREB3L1 premature stop codon in first case of recessive osteogenesis imperfecta associated with OASIS-deficiency to survive infancy". Bone. 114: 268–277. doi:10.1016/j.bone.2018.06.019. PMID 29936144. S2CID 49406925. Archived from the original on 29 August 2021. Retrieved 29 August 2021.
- Keller RB, Tran TT, Pyott SM, Pepin MG, Savarirayan R, McGillivray G, et al. (April 2018). "Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively". Genetics in Medicine. 20 (4): 411–419. doi:10.1038/gim.2017.115. PMC 5816725. PMID 28817112.
- Mendoza-Londono R, Fahiminiya S, Majewski J, Tétreault M, Nadaf J, Kannu P, et al. (June 2015). "Recessive osteogenesis imperfecta caused by missense mutations in SPARC". American Journal of Human Genetics. 96 (6): 979–985. doi:10.1016/j.ajhg.2015.04.021. PMC 4457955. PMID 26027498.
- Doyard M, Bacrot S, Huber C, Di Rocco M, Goldenberg A, Aglan MS, et al. (April 2018). "FAM46A mutations are responsible for autosomal recessive osteogenesis imperfecta". Journal of Medical Genetics. The BMJ. 55 (4): 278–284. doi:10.1136/jmedgenet-2017-104999. PMID 29358272. S2CID 4283476. Archived from the original on 29 August 2021. Retrieved 29 August 2021.
- Lindert U, Cabral WA, Ausavarat S, Tongkobpetch S, Ludin K, Barnes AM, et al. (July 2016). "MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta". Nature Communications. 7 (1): 11920. Bibcode:2016NatCo...711920L. doi:10.1038/ncomms11920. PMC 4935805. PMID 27380894.
- Moosa S, Yamamoto GL, Garbes L, Keupp K, Beleza-Meireles A, Moreno CA, et al. (October 2019). "Autosomal-Recessive Mutations in MESD Cause Osteogenesis Imperfecta". American Journal of Human Genetics. 105 (4): 836–843. doi:10.1016/j.ajhg.2019.08.008. PMC 6817720. PMID 31564437.
- Byers PH, Pyott SM (15 December 2012). "Recessively Inherited Forms of Osteogenesis Imperfecta". Annual Review of Genetics. 46 (1): 475–497. doi:10.1146/annurev-genet-110711-155608. ISSN 0066-4197. PMID 23145505.
- Ho Duy B, Zhytnik L, Maasalu K, Kändla I, Prans E, Reimann E, et al. (August 2016). "Mutation analysis of the COL1A1 and COL1A2 genes in Vietnamese patients with osteogenesis imperfecta". Human Genomics. 10 (1): 27. doi:10.1186/s40246-016-0083-1. PMC 4983065. PMID 27519266.
- Palomo T, Vilaça T, Lazaretti-Castro M (December 2017). "Osteogenesis imperfecta: diagnosis and treatment". Current Opinion in Endocrinology, Diabetes and Obesity. 24 (6): 381–388. doi:10.1097/MED.0000000000000367. PMID 28863000. S2CID 4555427.
- Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B (18 July 2014). "What is new in genetics and osteogenesis imperfecta classification?". Jornal de Pediatria. 90 (6): 536–541. doi:10.1016/j.jped.2014.05.003. PMID 25046257.
- Forlino A, Cabral WA, Barnes AM, Marini JC (June 2011). "New perspectives on osteogenesis imperfecta". Nature Reviews. Endocrinology. 7 (9): 540–557. doi:10.1038/nrendo.2011.81. PMC 3443407. PMID 21670757.
- Koromani F, Trajanoska K, Rivadeneira F, Oei L (4 June 2019). "Recent Advances in the Genetics of Fractures in Osteoporosis". Frontiers in Endocrinology. 10: 337. doi:10.3389/fendo.2019.00337. PMC 6559287. PMID 31231309.
- Rohrbach M, Giunta C (August 2012). "Recessive osteogenesis imperfecta: clinical, radiological, and molecular findings". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 160C (3): 175–189. doi:10.1002/ajmg.c.31334. PMID 22791419. S2CID 28592112.
- Marini JC, Reich A, Smith SM (August 2014). "Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation". Current Opinion in Pediatrics. 26 (4): 500–507. doi:10.1097/MOP.0000000000000117. PMC 4183132. PMID 25007323.
- Hanagata N (March 2016). "IFITM5 mutations and osteogenesis imperfecta". Journal of Bone and Mineral Metabolism. 34 (2): 123–131. doi:10.1007/s00774-015-0667-1. PMID 26031935. S2CID 3173191.
- Pyott SM, Pepin MG, Schwarze U, Yang K, Smith G, Byers PH (February 2011). "Recurrence of perinatal lethal osteogenesis imperfecta in sibships: parsing the risk between parental mosaicism for dominant mutations and autosomal recessive inheritance". Genetics in Medicine. 13 (2): 125–130. doi:10.1097/GIM.0b013e318202e0f6. PMID 21239989.
- Rauch F, Glorieux FH (April 2004). "Osteogenesis imperfecta". Lancet. 363 (9418): 1377–1385. doi:10.1016/S0140-6736(04)16051-0. PMID 15110498. S2CID 24081895.
- Gautieri A, Uzel S, Vesentini S, Redaelli A, Buehler MJ (August 2009). "Molecular and mesoscale mechanisms of osteogenesis imperfecta disease in collagen fibrils". Biophysical Journal. 97 (3): 857–865. Bibcode:2009BpJ....97..857G. doi:10.1016/j.bpj.2009.04.059. PMC 2718154. PMID 19651044.
- Xiao J, Cheng H, Silva T, Baum J, Brodsky B (December 2011). "Osteogenesis imperfecta missense mutations in collagen: structural consequences of a glycine to alanine replacement at a highly charged site". Biochemistry. 50 (50): 10771–10780. doi:10.1021/bi201476a. PMC 3292618. PMID 22054507.
- Nijhuis WH, Eastwood DM, Allgrove J, Hvid I, Weinans HH, Bank RA, Sakkers RJ (February 2019). "Current concepts in osteogenesis imperfecta: bone structure, biomechanics and medical management". Journal of Children's Orthopaedics. 13 (1): 1–11. doi:10.1302/1863-2548.13.180190. PMC 6376438. PMID 30838070.
- El-Sobky TA, Shawky RM, Sakr HM, Elsayed SM, Elsayed NS, Ragheb SG, Gamal R (15 November 2017). "A systematized approach to radiographic assessment of commonly seen genetic bone diseases in children: A pictorial review". J Musculoskelet Surg Res. 1 (2): 25. doi:10.4103/jmsr.jmsr_28_17. S2CID 79825711.
- Jameson JR, Albert CI, Busse B, Smith PA, Harris GF (29 March 2013). "3D micron-scale imaging of the cortical bone canal network in human osteogenesis imperfecta (OI)". In Weaver JB, Molthen RC (eds.). Medical Imaging 2013: Biomedical Applications in Molecular, Structural, and Functional Imaging. Vol. 8672. International Society for Optics and Photonics. pp. 86721L. doi:10.1117/12.2007209. S2CID 13876569. Archived from the original on 7 June 2020. Retrieved 26 August 2020.
- Westgren M, Götherström C (September 2015). "Stem cell transplantation before birth - a realistic option for treatment of osteogenesis imperfecta?". Prenatal Diagnosis. 35 (9): 827–832. doi:10.1002/pd.4611. PMID 25962526. S2CID 10640427.
- Pepin MG, Byers PH (December 2015). "What every clinical geneticist should know about testing for osteogenesis imperfecta in suspected child abuse cases". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 169 (4): 307–313. doi:10.1002/ajmg.c.31459. PMID 26566591. S2CID 26045033.
- Castaneda R (25 October 2005). "Parents Get $1 Million in False Abuse Case". Washington Post. ISSN 0190-8286. Retrieved 4 February 2022.
- "What Is Osteogenesis Imperfecta? Fast Facts: An Easy-to-Read Series of Publications for the Public". NIAMS. November 2014. Archived from the original on 18 October 2016. Retrieved 15 October 2016.
- Swezey T, Reeve BB, Hart TS, Floor MK, Dollar CM, Gillies AP, Tosi LL (February 2019). "Incorporating the patient perspective in the study of rare bone disease: insights from the osteogenesis imperfecta community". Osteoporosis International. 30 (2): 507–511. doi:10.1007/s00198-018-4690-7. PMC 6449303. PMID 30191258.
- Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R (October 1998). "Cyclic administration of pamidronate in children with severe osteogenesis imperfecta". The New England Journal of Medicine. 339 (14): 947–952. doi:10.1056/NEJM199810013391402. PMID 9753709. S2CID 19316414.Free full text
- DiMeglio LA, Peacock M (January 2006). "Two-year clinical trial of oral alendronate versus intravenous pamidronate in children with osteogenesis imperfecta". Journal of Bone and Mineral Research. 21 (1): 132–140. doi:10.1359/JBMR.051006. PMID 16355282. S2CID 12996685.
- Bishop N, Adami S, Ahmed SF, Antón J, Arundel P, Burren CP, et al. (October 2013). "Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial". Lancet. 382 (9902): 1424–1432. doi:10.1016/S0140-6736(13)61091-0. PMID 23927913. S2CID 25559791.
- Ward LM, Rauch F (October 2013). "Oral bisphosphonates for paediatric osteogenesis imperfecta?". Lancet. 382 (9902): 1388–1389. doi:10.1016/S0140-6736(13)61531-7. PMID 23927912. S2CID 5872511.
- Meena BL, Panigrahi I, Marwaha RK (1 October 2014). "PO-0077 Vitamin D Deficiency In Children With Osteogenesis Imperfecta". Archives of Disease in Childhood. 99 (Suppl 2): A275. doi:10.1136/archdischild-2014-307384.747. ISSN 0003-9888. S2CID 72491188. Archived from the original on 18 September 2021. Retrieved 18 September 2021.
- Edouard T, Glorieux FH, Rauch F (October 2011). "Predictors and correlates of vitamin D status in children and adolescents with osteogenesis imperfecta". The Journal of Clinical Endocrinology and Metabolism. 96 (10): 3193–3198. doi:10.1210/jc.2011-1480. PMID 21832107.
- Zambrano MB, Brizola E, Pinheiro B, Vanz AP, Mello ED, Félix TM (18 May 2016). "Study of the Determinants of Vitamin D Status in Pediatric Patients With Osteogenesis Imperfecta". Journal of the American College of Nutrition. 35 (4): 339–345. doi:10.1080/07315724.2015.1057776. PMID 26709914. S2CID 45930763.
- Rothschild L, Goeller JK, Voronov P, Barabanova A, Smith P (November 2018). "Anesthesia in children with osteogenesis imperfecta: Retrospective chart review of 83 patients and 205 anesthetics over 7 years". Pediatric Anesthesia. 28 (11): 1050–1058. doi:10.1111/pan.13504. PMID 30295359. S2CID 52929810. Archived from the original on 29 August 2021. Retrieved 29 August 2021.
- Houck JP (2014). "Osteogenesis imperfecta". In Houck JP, Hache M, Sun LS (eds.). Handbook of Pediatric Anesthesia. New York: McGraw-Hill Education. ISBN 9780071769358. Archived from the original on 29 August 2021. Retrieved 29 August 2021.
- Goeller JK, Rothschild L (2020), Kruse RW (ed.), "Anesthetic and Post-operative Pain Management" (ePub), Osteogenesis Imperfecta: A Case-Based Guide to Surgical Decision-Making and Care, Springer International Publishing, pp. 111–125, doi:10.1007/978-3-030-42527-2_8, ISBN 978-3-030-42527-2, S2CID 219515776
- Behera P, Santoshi JA, Geevarughese NM, Meena UK, Selvanayagam R (28 February 2020). "Dislodgement of Telescopic Nail from the Epiphysis: A Case Report with an Analysis of Probable Mechanism". Cureus. 12 (2): e7130. doi:10.7759/cureus.7130. ISSN 2168-8184. PMC 7105006. PMID 32257675.
- "Financial Assistance". Shriners Hospitals for Children. Archived from the original on 3 March 2021. Retrieved 21 August 2021.
Shriners Hospitals for Children is committed to providing specialized care to children with orthopaedic conditions [...] regardless of the families' ability to pay.
- "A Leader in the Treatment of Osteogensis Imperfecta (OI)". Shriners International. Archived from the original on 28 September 2007. Retrieved 5 July 2007.
- Sofield HA, Millar EA (1 December 1959). "Fragmentation, Realignment, and Intramedullary Rod Fixation of Deformities of the Long Bones in Children: A Ten-Year Appraisal". The Journal of Bone and Joint Surgery. 41 (8): 1371–1391. doi:10.2106/00004623-195941080-00001. ISSN 0021-9355.
- "The Fassier-Duval Telescopic IM System". Pega Medical. Laval, Quebec. Archived from the original on 15 August 2021. Retrieved 15 August 2021.
- Sterian A, Balanescu R, Barbilian A, Ulici A (2015). "Osteosynthesis in Osteogenesis Imperfecta, telescopic versus non-telescopic nailing". Journal of Medicine and Life. 8 (4): 563–565. PMC 4656972. PMID 26664490.
- Frassier F, Duval P, Paley D (2021). "Fassier–Duval Telescopic IM System Surgical Technique" (PDF). Pega Medical. Laval, Quebec. FD-ST-EN Revision K. Archived (PDF) from the original on 15 August 2021. Retrieved 15 August 2021.
- Belyea CM, Knox JB (1 January 2020). "Spinal fusion in children with osteogenesis imperfecta: A nationwide retrospective comparative cohort study over a 12-year period". Current Orthopaedic Practice. 31 (1): 72–75. doi:10.1097/BCO.0000000000000805. ISSN 1940-7041. S2CID 209229412.
- Zani A, Ford-Adams M, Ratcliff M, Bevan D, Inge TH, Desai A (January 2017). "Weight loss surgery improves quality of life in pediatric patients with osteogenesis imperfecta". Surgery for Obesity and Related Diseases. Elsevier Inc. 13 (1): 41–44. doi:10.1016/j.soard.2015.11.029. PMC 5965274. PMID 26948942.
- Semler O, Fricke O, Vezyroglou K, Stark C, Schoenau E (1 January 2007). "Preliminary results on the mobility after whole body vibration in immobilized children and adolescents". Journal of Musculoskeletal & Neuronal Interactions. 7 (1): 77–81. PMID 17396011. Archived from the original on 18 August 2021. Retrieved 18 August 2021.
- Biria M, Abbas FM, Mozaffar S, Ahmadi R (July 2012). "Dentinogenesis imperfecta associated with osteogenesis imperfecta". Dental Research Journal. 9 (4): 489–494. PMC 3491340. PMID 23162594.
- Plieth J (18 December 2020). "Recycling pays off for Mereo". Evaluate Vantage. Archived from the original on 18 September 2021. Retrieved 18 September 2021.
- Delgado-Calle J, Sato AY, Bellido T (March 2017). "Role and mechanism of action of sclerostin in bone". Bone. 96: 29–37. doi:10.1016/j.bone.2016.10.007. PMC 5328835. PMID 27742498.
- Glorieux FH, Devogelaer JP, Durigova M, Goemaere S, Hemsley S, Jakob F, et al. (July 2017). "BPS804 Anti-Sclerostin Antibody in Adults With Moderate Osteogenesis Imperfecta: Results of a Randomized Phase 2a Trial". Journal of Bone and Mineral Research. 32 (7): 1496–1504. doi:10.1002/jbmr.3143. PMID 28370407. S2CID 4432023.
- "Ultragenyx and Mereo BioPharma Announce Collaboration and License Agreement for Setrusumab in Osteogenesis Imperfecta". Ultragenyx Pharmaceutical Inc. 17 December 2020. Archived from the original on 18 September 2021. Retrieved 18 September 2021.
- "A Study in Adult Patients With Type I, III or IV Osteogenesis Imperfecta Treated With BPS804 (ASTEROID)". ClinicalTrials.gov. United States National Library of Medicine. 1 April 2021. NCT03118570. Retrieved 18 September 2021.
- Cooper M, Gregorek L (25 November 2019). "Setrusumab ready to advance into pivotal studies". Trinity Delta. Archived from the original on 18 September 2021. Retrieved 18 September 2021.
- Hardy A (24 September 2020). "Mereo setrusumab gets FDA rare pediatric disease designation for osteogenesis imperfecta". BioTuesdays. Archived from the original on 18 September 2021. Retrieved 18 September 2021.
- "Study to Evaluate Romosozumab in Children and Adolescents With Osteogenesis Imperfecta". ClinicalTrials.gov. United States National Library of Medicine. 23 July 2021. NCT04545554. Archived from the original on 18 September 2021. Retrieved 18 September 2021.
- Out of Ignorance (2 September 2022). "Ultragenyx Stock: Downward Trajectory Likely to Continue". Seeking Alpha. Retrieved 24 September 2022.
- Ultragenyx Pharmaceutical Inc (15 September 2022). "A Randomized, Double-Blind, Placebo-Controlled, Phase 2/3 Study to Assess the Efficacy and Safety of Setrusumab in Subjects With Osteogenesis Imperfecta". Mereo BioPharma.
{{cite journal}}: Cite journal requires|journal=(help) - Green R (11 March 2012). "Brittle Bone Disease Forced Mother Into 'No Easy Choice'". Hartford Courant. Retrieved 16 September 2021.
- Hopper T (6 February 2013). "'Incompatible with life': Mother's account of her difficult decision to request a late-term abortion sheds light on 'live-born' debate". National Post. Retrieved 16 September 2021.
- Paterson CR, Ogston SA, Henry RM (February 1996). "Life expectancy in osteogenesis imperfecta". BMJ. 312 (7027): 351. doi:10.1136/bmj.312.7027.351. PMC 2350292. PMID 8611834.
- Plotkin H (29 February 2016). "Genetics of Osteogenesis Imperfecta". Archived from the original on 30 December 2010.
- "2.4: The Frequency of Bone Disease". Bone Health and Osteoporosis: A Report of the Surgeon General (PDF). Rockville, Maryland: Office of the Surgeon General of the United States, U.S. Department of Health and Human Services. 2004. p. 68. WE 225 B71259 2004.
- "Osteogenesis Imperfecta Panel". University of Nebraska Medical Center. Archived from the original on 30 July 2019. Retrieved 30 July 2019.
- Abali S, Arman A, Atay Z, Bereket A, Bas S, Haliloglu B, et al. (19 August 2016). "Frequency of Recessive Osteogenesis Imperfecta in a Turkish Cohort and Genetic Causes". ESPE Abstracts. Bioscientifica. 86: 131. Archived from the original on 26 August 2021. Retrieved 26 August 2021.
- Lowenstein EJ (May 2009). "Osteogenesis imperfecta in a 3,000-year-old mummy". Child's Nervous System. 25 (5): 515–516. doi:10.1007/s00381-009-0817-7. PMID 19212769.
- Tainmont J (2007). "History of osteogenesis imperfecta or brittle bone disease: a few stops on a road 3000 years long". B-Ent. 3 (3): 157–173. PMID 17970442. Archived from the original on 29 August 2021. Retrieved 29 August 2021.
- Groeneveld E (12 November 2018). "Ivar the Boneless". World History Encyclopedia. Ancient History Encyclopedia Limited. Archived from the original on 21 August 2021. Retrieved 21 August 2021.
- Lewis R, Lotha G, Gaur A (8 January 2019). "Ivar the Boneless". Encyclopedia Britannica. Archived from the original on 24 June 2021. Retrieved 21 August 2021.
- Malebranche N (1688). (in French) (1842 ed.). p. 119 – via Wikisource.
- Edelu B, Ndu I, Asinobi I, Obu H, Adimora G (March 2014). "Osteogenesis imperfecta: a case report and review of literature". Annals of Medical and Health Sciences Research. Nigerian Medical Association. 4 (Suppl 1): S1–S5. doi:10.4103/2141-9248.131683. PMC 4083720. PMID 25031897.
- Hektoen L (May 1903). "Anatomical Study of a Short-Limbed Dwarf, With Special Reference To Osteogenesis Imperfecta and Chondrodystrophia Foetalis". The American Journal of the Medical Sciences. J. B. Lippincott & Co. 125 (5): 751–769. doi:10.1097/00000441-190305000-00001. ISBN 978-0-243-38392-4. S2CID 71836609. Archived from the original on 16 August 2021. Retrieved 16 August 2021.
- Adair-Dighton CA (1912). "Four generations of blue sclerotics". The Ophthalmoscope. London: George Pulman & Sons, Ltd. 10: 188–189. OCLC 1049881577 – via Internet Archive.
- Vrolik W (1849). Tabulae ad illustrandam embryogenesin hominis et mammalium, tam naturalem quam abnormem — De Vrucht van den Mensch en van de Zoogdieren afgebeeld en beschreven in hare regelmatige en onregelmatige Ontwikkeling [Illustrations of human and mammalian embryogenesis, both normal and abnormal] (in Latin and Dutch). Amsterdam: G.M.P. Londonck. pp. 562–566. OCLC 489920691. Archived from the original on 29 August 2021. Retrieved 29 August 2021.
- Baljet B (January 2002). "Aspects of the history of osteogenesis imperfecta (Vrolik's syndrome)". Annals of Anatomy - Anatomischer Anzeiger. 184 (1): 1–7. doi:10.1016/S0940-9602(02)80023-1. PMID 11876477. Archived from the original on 12 June 2018. Retrieved 29 August 2021.
- Baljet B (2002). "Willem Vrolik and "his" Syndrome". In Rauschmann MA, Thomann KD, Zichner, L (eds.). Deutsches Orthopädisches Geschichts und Forschungsmuseum. Vol. 4. Heidelberg: Verlag Theodor Steinkopff. pp. 133–144. doi:10.1007/978-3-642-57510-5_15. ISBN 978-3-642-57510-5. Archived from the original on 8 June 2018. Retrieved 29 August 2021.
- Looser E (1906). "Zur Kenntnis der Osteogenesis Imperfecta Congenita et Tarda (sogenannte idiopathische Osteopsatyrosis)" [On awareness of osteogenesis imperfecta congenita and tarda (so-called "idiopathic osteopsatyrosis")]. Mitteilungen aus den Grenzgebieten der Medizin und Chirurgie (Dispatches from the Frontier of Medicine and Surgery) (in German). 15: 161–207. Archived from the original on 21 August 2021. Retrieved 21 August 2021.
- synd/1743 at Who Named It?
- Rasmussen C, Danks A (2010). Double helix, double joy: David Danks, the father of clinical genetics in Australia. Carlton, Victoria: Melbourne University Press. p. 116. ISBN 978-0-522-85799-3. OCLC 906592657. Archived from the original on 28 August 2021. Retrieved 28 August 2021.
- Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, et al. (May 2011). "Nosology and classification of genetic skeletal disorders: 2010 revision". American Journal of Medical Genetics. Part A. 155A (5): 943–968. doi:10.1002/ajmg.a.33909. PMC 3166781. PMID 21438135.
- "Osteogenesis Imperfecta (OI) in Dachshunds". UC Davis Veterinary Genetics Laboratory. UC Davis School of Veterinary Medicine. Archived from the original on 28 August 2021. Retrieved 28 August 2021.
- "Osteogenesis Imperfecta (OI Dachsund)". Center for Animal Genetics. Generatio GmbH. Archived from the original on 8 November 2018. Retrieved 7 November 2018.
- Enderli TA, Burtch SR, Templet JN, Carriero A (September 2016). "Animal models of osteogenesis imperfecta: applications in clinical research". Orthopedic Research and Reviews. 8: 41–55. doi:10.2147/ORR.S85198. PMC 6209373. PMID 30774469.
- Saban J, Zussman MA, Havey R, Patwardhan AG, Schneider GB, King D (December 1996). "Heterozygous oim mice exhibit a mild form of osteogenesis imperfecta". Bone. 19 (6): 575–579. doi:10.1016/S8756-3282(96)00305-5. PMID 8968022. Archived from the original on 30 June 2018. Retrieved 28 August 2021.
{kind=link}
{kind=link}
External links
- "Osteogenesis Imperfecta Overview". NIH Osteoporosis and Related Bone Diseases — National Resource Center. National Institutes of Health, U.S. Department of Health and Human Services.