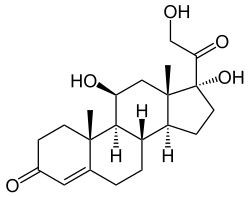
Cortisol
 | |
 | |
| Names | |
|---|---|
| IUPAC name
11β,17α,21-Trihydroxypregn-4-ene-3,20-dione | |
| Preferred IUPAC name
(1R,3aS,3bS,9aR,9bS,11aS)-1,10-Dihydroxy-1-(hydroxyacetyl)-9a,11a-dimethyl-1,2,3,3a,3b,4,5,8,9,9a,9b,10,11,11a-tetradecahydro-7H-cyclopenta[a]phenanthen-7-one | |
| Identifiers | |
3D model (JSmol) |
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank | |
| ECHA InfoCard | 100.000.019 |
| KEGG | |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C21H30O5 | |
| Molar mass | 362.460 g/mol |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |
Cortisol is a steroid hormone, in the glucocorticoid class of hormones. When used as a medication, it is known as hydrocortisone.
It is produced in many animals, mainly by the zona fasciculata of the adrenal cortex in the adrenal gland.[1] It is produced in other tissues in lower quantities.[2] It is released with a diurnal cycle and its release is increased in response to stress and low blood-glucose concentration. It functions to increase blood sugar through gluconeogenesis, to suppress the immune system, and to aid in the metabolism of fat, protein, and carbohydrates.[3] It also decreases bone formation.[4]
Health effects
Metabolism of glucose
In general, cortisol stimulates gluconeogenesis (the synthesis of 'new' glucose from non-carbohydrate sources, which occurs mainly in the liver, but also in the kidneys and small intestine under certain circumstances). The net effect is an increase in the concentration of glucose in the blood, further complemented by a decrease in the sensitivity of peripheral tissue to insulin, thus preventing this tissue from taking the glucose from the blood. Cortisol has a permissive effect on the actions of hormones that increase glucose production, such as glucagon and adrenaline.[5]
Cortisol also plays an important, but indirect, role in liver and muscle glycogenolysis (the breaking down of glycogen to glucose-1-phosphate and glucose) which occurs as a result of the action of glucagon and adrenaline. Additionally, cortisol facilitates the activation of glycogen phosphorylase, which is necessary for adrenaline to have an effect on glycogenolysis.[6][7]
Paradoxically, cortisol promotes not only gluconeogenesis in the liver, but also glycogenesis. Cortisol is thus better thought of as stimulating glucose/glycogen turnover in the liver.[8] This is in contrast to cortisol's effect in the skeletal muscle where glycogenolysis is promoted indirectly through catecholamines.[9]
Metabolism of proteins and lipids
Elevated levels of cortisol, if prolonged, can lead to proteolysis (breakdown of proteins) and muscle wasting.[10] The reason for proteolysis is to provide the relevant tissue with a feedstock for gluconeogenesis; see glucogenic amino acids.[5] The effects of cortisol on lipid metabolism are more complicated since lipogenesis is observed in patients with chronic, raised circulating glucocorticoid (i.e. cortisol) levels,[5] although an acute increase in circulating cortisol promotes lipolysis.[11] The usual explanation to account for this apparent discrepancy is that the raised blood glucose concentration (through the action of cortisol) will stimulate insulin release. Insulin stimulates lipogenesis, so this is an indirect consequence of the raised cortisol concentration in the blood but it will only occur over a longer time scale.
Immune response
Cortisol prevents the release of substances in the body that cause inflammation. It is used to treat conditions resulting from overactivity of the B-cell-mediated antibody response. Examples include inflammatory and rheumatoid diseases, as well as allergies. Low-dose topical hydrocortisone, available as a nonprescription medicine in some countries, is used to treat skin problems such as rashes and eczema.
Cortisol inhibits production of interleukin 12 (IL-12), interferon gamma (IFN-gamma), IFN-alpha, and tumor necrosis factor alpha (TNF-alpha) by antigen-presenting cells (APCs) and T helper cells (Th1 cells), but upregulates interleukin 4, interleukin 10, and interleukin 13 by Th2 cells. This results in a shift toward a Th2 immune response rather than general immunosuppression. The activation of the stress system (and resulting increase in cortisol and Th2 shift) seen during an infection is believed to be a protective mechanism which prevents an over-activation of the inflammatory response.[12]
Cortisol can weaken the activity of the immune system. It prevents proliferation of T-cells by rendering the interleukin-2 producer T-cells unresponsive to interleukin-1, and unable to produce the T-cell growth factor IL-2. Cortisol downregulates the expression of the IL2 receptor IL-2R on the surface of the helper T-cell which is necessary to induce a Th1 'cellular' immune response, thus favoring a shift towards Th2 dominance and the release of the cytokines listed above which results in Th2 dominance and favors the 'humoral' B-cell mediated antibody immune response).[13]
Cortisol also has a negative-feedback effect on IL-1.[14] The way this negative feedback works is that an immune stressor causes peripheral immune cells to release IL-1 and other other cytokines such as IL-6 and TNF-alpha. These cytokines stimulate the hypothalamus, causing it to release corticotropin-releasing hormone (CRH). CRH in turn stimulates the production of adrenocorticotropic hormone (ACTH) among other things in the adrenal gland, which (among other things) increases production of cortisol. Cortisol then closes the loop as it inhibits TNF-alpha production in immune cells and makes them less responsive to IL-1.[15]
Through this system, as long as an immune stressor is small, the response will be regulated to the correct level. Like a thermostat controlling a heater, the hypothalamus uses cortisol to turn off the heat once the production of cortisol matches the stress induced on the immune system. But in a severe infection or in a situation where the immune system is overly sensitized to an antigen (such as in allergic reactions) or there is a massive flood of antigens (as can happen with endotoxic bacteria) the correct set point might never be reached. Also because of downregulation of Th1 immunity by cortisol and other signaling molecules, certain types of infection, (notably Mycobacterium tuberculosis) can trick the body into getting locked in the wrong mode of attack, using an antibody-mediated humoral response when a cellular response is needed.
Lymphocytes are the antibody-producing cells of the body, and are thus the main agents of humoral immunity. A larger number of lymphocytes in the lymph nodes, bone marrow, and skin means the body is increasing its humoral immune response. Lymphocytes release antibodies into the bloodstream. These antibodies lower infection through three main pathways: neutralization, opsonization, and complement activation. Antibodies neutralize pathogens by binding to surface adhering proteins, keeping pathogens from binding to host cells. In opsonization, antibodies bind to the pathogen and create a target for phagocytic immune cells to find and latch onto, allowing them to destroy the pathogen more easily. Finally antibodies can also activate complement molecules which can combine in various ways to promote opsonization or even act directly to lyse a bacteria. There are many different kinds of antibody and their production is highly complex, involving several types of lymphocyte, but in general lymphocytes and other antibody regulating and producing cells will migrate to the lymph nodes to aid in the release of these antibodies into the bloodstream.[16]
Rapid administration of corticosterone (the endogenous type I and type II receptor agonist) or RU28362 (a specific type II receptor agonist) to adrenalectomized animals induced changes in leukocyte distribution.
On the other side of things, there are natural killer cells; these cells are equipped with the heavy artillery needed to take down larger in size threats like bacteria, parasites, and tumor cells. A separate study[17] found that cortisol effectively disarmed natural killer cells, downregulating the expression of their natural cytotoxicity receptors. Interestingly, prolactin has the opposite effect. It increases the expression of cytotoxicity receptors on natural killer cells, increasing their firepower.
Cortisol stimulates many copper enzymes (often to 50% of their total potential), including lysyl oxidase, an enzyme that cross-links collagen and elastin. Especially valuable for immune response is cortisol's stimulation of the superoxide dismutase,[18] since this copper enzyme is almost certainly used by the body to permit superoxides to poison bacteria.
Other effects
Glucose
Cortisol counteracts insulin, contributes to hyperglycemia by stimulating gluconeogenesis[19] and inhibits the peripheral use of glucose (insulin resistance)[19] by decreasing the translocation of glucose transporters (especially GLUT4) to the cell membrane.[20] Cortisol also increases glycogen synthesis (glycogenesis) in the liver, storing glucose in easily accessible form.[21] The permissive effect of cortisol on insulin action in liver glycogenesis is observed in hepatocyte culture in the laboratory, although the mechanism for this is unknown.
Bone and collagen
Cortisol reduces bone formation,[4] favoring long-term development of osteoporosis (progressive bone disease). The mechanism behind this is two-fold: cortisol stimulates the production of RANKL by osteoblasts which stimulates, through binding to RANK receptors, the activity of osteoclasts – cells responsible for calcium resorption from bone – and also inhibits the production of osteoprotegerin (OPG) which acts as a decoy receptor and captures some RANKL before it can activate the osteoclasts through RANK.[5] In other words, when RANKL binds to OPG, no response occurs as opposed to the binding to RANK which leads to the activation of osteoclasts.
It transports potassium out of cells in exchange for an equal number of sodium ions (see above).[22] This can trigger the hyperkalemia of metabolic shock from surgery. Cortisol also reduces calcium absorption in the intestine.[23] Cortisol down-regulates the synthesis of collagen.[24]
Amino acid
Cortisol raises the free amino acids in the serum by inhibiting collagen formation, decreasing amino acid uptake by muscle, and inhibiting protein synthesis.[25] Cortisol (as opticortinol) may inversely inhibit IgA precursor cells in the intestines of calves.[26] Cortisol also inhibits IgA in serum, as it does IgM; however, it is not shown to inhibit IgE.[27]
Electrolyte balance
Cortisol decreases glomerular filtration rate, and renal plasma flow from the kidneys thus increasing phosphate excretion, as well as increasing sodium and water retention and potassium excretion by acting on mineralocorticoid receptors. It also increases sodium and water absorption and potassium excretion in the intestines.[28]
Sodium
Cortisol promotes sodium absorption through the small intestine of mammals.[29] Sodium depletion, however, does not affect cortisol levels[30] so cortisol cannot be used to regulate serum sodium. Cortisol's original purpose may have been sodium transport. This hypothesis is supported by the fact that freshwater fish use cortisol to stimulate sodium inward, while saltwater fish have a cortisol-based system for expelling excess sodium.[31]
Potassium
A sodium load augments the intense potassium excretion by cortisol. Corticosterone is comparable to cortisol in this case.[32] For potassium to move out of the cell, cortisol moves an equal number of sodium ions into the cell.[22] This should make pH regulation much easier (unlike the normal potassium-deficiency situation, in which two sodium ions move in for each three potassium ions that move out—closer to the deoxycorticosterone effect).
Stomach and kidneys
Cortisol stimulates gastric-acid secretion.[33] Cortisol's only direct effect on the hydrogen-ion excretion of the kidneys is to stimulate the excretion of ammonium ions by deactivating the renal glutaminase enzyme.[34]
Memory
Cortisol works with adrenaline (epinephrine) to create memories of short-term emotional events; this is the proposed mechanism for storage of flash bulb memories, and may originate as a means to remember what to avoid in the future.[35] However, long-term exposure to cortisol damages cells in the hippocampus;[36] this damage results in impaired learning.
Diurnal cycles

Diurnal cycles of cortisol levels are found in humans.[6]
Stress
Sustained stress can lead to high levels of circulating cortisol (regarded as one of the more important of the several "stress hormones".[37]
Effects during pregnancy
During human pregnancy, increased fetal production of cortisol between weeks 30 and 32 initiates production of fetal lung pulmonary surfactant to promote maturation of the lungs. In fetal lambs, glucocorticoids (principally cortisol) increase after about day 130, with lung surfactant increasing greatly, in response, by about day 135,[38] and although lamb fetal cortisol is mostly of maternal origin during the first 122 days, 88% or more is of fetal origin by day 136 of gestation.[39] Although the timing of fetal cortisol concentration elevation in sheep may vary somewhat, it averages about 11.8 days before the onset of labor.[40] In several livestock species (e.g. cattle, sheep, goats, and pigs), the surge of fetal cortisol late in gestation triggers the onset of parturition by removing the progesterone block of cervical dilation and myometrial contraction. The mechanisms yielding this effect on progesterone differ among species. In the sheep, where progesterone sufficient for maintaining pregnancy is produced by the placenta after about day 70 of gestation,[41][42] the prepartum fetal cortisol surge induces placental enzymatic conversion of progesterone to estrogen. (The elevated level of estrogen stimulates prostaglandin secretion and oxytocin receptor development.)
Exposure of fetuses to cortisol during gestation can have a variety of developmental outcomes, including alterations in prenatal and postnatal growth patterns. In marmosets, a species of New World primates, pregnant females have varying levels of cortisol during gestation, both within and between females. Infants born to mothers with high gestational cortisol during the first trimester of pregnancy had lower rates of growth in body mass indices than infants born to mothers with low gestational cortisol (about 20% lower). However, postnatal growth rates in these high-cortisol infants were more rapid than low-cortisol infants later in postnatal periods, and complete catch-up in growth had occurred by 540 days of age. These results suggest that gestational exposure to cortisol in fetuses has important potential fetal programming effects on both pre and postnatal growth in primates.[43]
Synthesis and release
Cortisol is produced in the human body by the adrenal gland in the zona fasciculata,[1] the second of three layers comprising the adrenal cortex. The cortex forms the outer "bark" of each adrenal gland, situated atop the kidneys. The release of cortisol is controlled by the hypothalamus, a part of the brain. The secretion of corticotropin-releasing hormone by the hypothalamus[44] triggers cells in the neighboring anterior pituitary to secrete another hormone, the adrenocorticotropic hormone (ACTH), into the vascular system, through which blood carries it to the adrenal cortex. ACTH stimulates the synthesis of cortisol and other glucocorticoids, mineralocorticoid aldosterone, and dehydroepiandrosterone.[45]
Testing of individuals
Normal values indicated in the following tables pertain to humans (normal levels vary among species). Measured cortisol levels, and therefore reference ranges, depend on the sample type (blood or urine), analytical method used, and factors such as age and sex. Test results should, therefore, always be interpreted using the reference range from the laboratory that produced the result.
| Time | Lower limit | Upper limit | Unit |
|---|---|---|---|
| 09:00 am | 140[46] | 700[46] | nmol/L |
| 5[47] | 25[47] | μg/dL | |
| Midnight | 80[46] | 350[46] | nmol/L |
| 2.9[47] | 13[47] | μg/dL |
Using the molecular weight of 362.460 g/mole, the conversion factor from µg/dL to nmol/L is approximately 27.6; thus, 10 µg/dL is about 276 nmol/L.
| Lower limit | Upper limit | Unit |
|---|---|---|
| 28[48] or 30[49] | 280[48] or 490[49] | nmol/24h |
| 10[50] or 11[51] | 100[50] or 176[51] | µg/24 h |
Cortisol follows a circadian rhythm, and to accurately measure cortisol levels is best to test four times per day through saliva. An individual may have normal total cortisol but have a lower than normal level during a certain period of the day and a higher than normal level during a different period. Therefore, some scholars question the clinical utility of cortisol measurement.[52][53][54][55]
Cortisol is lipophilic, and is transported bound to transcortin (also known as corticosteroid-binding globulin) and albumin, while only a small part of the total serum cortisol is unbound and has biological activity.[56] Serum cortisol assays measures total cortisol, and its results may be misleading for patients with altered serum protein concentrations. The salivary cortisol test avoids this problem because only free cortisol can pass through the salivary barrier. Transcortin particles are too large to pass through this barrier.
Automated immunoassays lack specificity and show significant cross-reactivity due to interactions with structural analogs of cortisol, and show differences between assays. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) can improve specificity and sensitivity.[57]
Disorders of cortisol production
Some medical disorders are related to abnormal cortisol production, such as:
- Primary hypercortisolism (Cushing's syndrome): excessive levels of cortisol[58]
- Primary hypocortisolism (Addison's disease, Nelson's syndrome): insufficient levels of cortisol
- Secondary hypocortisolism (pituitary tumor, Sheehan's syndrome)
Regulation
The primary control of cortisol is the pituitary gland peptide, ACTH, which probably controls cortisol by controlling the movement of calcium into the cortisol-secreting target cells.[61] ACTH is in turn controlled by the hypothalamic peptide corticotropin-releasing hormone (CRH), which is under nervous control. CRH acts synergistically with arginine vasopressin, angiotensin II, and epinephrine.[62] (In swine, which do not produce arginine vasopressin, lysine vasopressin acts synergistically with CRH.[63])
When activated macrophages start to secrete IL-1, which synergistically with CRH increases ACTH,[14] T-cells also secrete glucosteroid response modifying factor (GRMF), as well as IL-1; both increase the amount of cortisol required to inhibit almost all the immune cells.[64] Immune cells then assume their own regulation, but at a higher cortisol setpoint. The increase in cortisol in diarrheic calves is minimal over healthy calves, however, and falls over time.[65] The cells do not lose all their fight-or-flight override because of interleukin-1's synergism with CRH. Cortisol even has a negative feedback effect on interleukin-1[14]—especially useful to treat diseases that force the hypothalamus to secrete too much CRH, such as those caused by endotoxic bacteria. The suppressor immune cells are not affected by GRMF,[64] so the immune cells' effective setpoint may be even higher than the setpoint for physiological processes. GRMF affects primarily the liver (rather than the kidneys) for some physiological processes.[66]
High-potassium media (which stimulates aldosterone secretion in vitro) also stimulate cortisol secretion from the fasciculata zone of canine adrenals[67][68] — unlike corticosterone, upon which potassium has no effect.[69]
Potassium loading also increases ACTH and cortisol in humans.[70] This is probably the reason why potassium deficiency causes cortisol to decline (as mentioned) and causes a decrease in conversion of 11-deoxycortisol to cortisol.[71] This may also have a role in rheumatoid-arthritis pain; cell potassium is always low in RA.[72]
Ascorbic acid presence, particularly in high doses has also been shown to mediate response to psychological stress and speed the decrease of the levels of circulating cortisol in the body post-stress. This can be evidenced through a decrease in systolic and diastolic blood pressures and decreased salivary cortisol levels after treatment with ascorbic acid.[73]
Factors increasing cortisol levels
- Viral infections increase cortisol levels through activation of the HPA axis by cytokines.[74]
- Intense (high VO2 max) or prolonged aerobic exercise transiently increases cortisol levels to increase gluconeogenesis and maintain blood glucose;[75] however, cortisol declines to normal levels after eating (i.e., restoring a neutral energy balance)[76]
- Severe trauma or stressful events can elevate cortisol levels in the blood for prolonged periods.[77]
- Low-carbohydrate diets cause a short-term increase in resting cortisol (~3 weeks), and increase the cortisol response to aerobic exercise in the short- and long-term.[78]
Biochemistry
Biosynthesis

Cortisol is synthesized from cholesterol. Synthesis takes place in the zona fasciculata of the adrenal cortex. (The name cortisol is derived from cortex.) While the adrenal cortex also produces aldosterone (in the zona glomerulosa) and some sex hormones (in the zona reticularis), cortisol is its main secretion in humans and several other species. (However, in cattle, corticosterone levels may approach[80] or exceed[6] cortisol levels.). The medulla of the adrenal gland lies under the cortex, mainly secreting the catecholamines adrenaline (epinephrine) and noradrenaline (norepinephrine) under sympathetic stimulation.
The synthesis of cortisol in the adrenal gland is stimulated by the anterior lobe of the pituitary gland with ACTH; ACTH production is, in turn, stimulated by CRH, which is released by the hypothalamus. ACTH increases the concentration of cholesterol in the inner mitochondrial membrane, via regulation of the steroidogenic acute regulatory protein. It also stimulates the main rate-limiting step in cortisol synthesis, in which cholesterol is converted to pregnenolone and catalyzed by Cytochrome P450SCC (side-chain cleavage enzyme).[81]
11beta-hydroxysteroid dehydrogenases
Cortisol is metabolized reversibly to cortisone[82] by the 11-beta hydroxysteroid dehydrogenase system (11-beta HSD), which consists of two enzymes: 11-beta HSD1 and 11-beta HSD2. The metabolism of cortisol to cortisone involves oxidation of the hydroxyl group at the 11-beta position.[83]
- 11-beta HSD1 uses the cofactor NADPH to convert biologically inert cortisone to biologically active cortisol
- 11-beta HSD2 uses the cofactor NAD+ to convert cortisol to cortisone
Overall, the net effect is that 11-beta HSD1 serves to increase the local concentrations of biologically active cortisol in a given tissue; 11-beta HSD2 serves to decrease local concentrations of biologically active cortisol. If hexose-6-phosphate dehydrogenase (H6PDH) is present, the equilibrium can favor the activity of 11-beta HSD1. H6PDH regenerates NADPH, which increases the activity of 11-beta HSD1, and decreases the activity of 11-beta HSD2.[84]
An alteration in 11-beta HSD1 has been suggested to play a role in the pathogenesis of obesity, hypertension, and insulin resistance known as metabolic syndrome.[85]
An alteration in 11-beta HSD2 has been implicated in essential hypertension and is known to lead to the syndrome of apparent mineralocorticoid excess (SAME).
A-ring reductases (5alpha- and 5beta-reductases)
Cortisol is also metabolized irreversibly into 5-alpha tetrahydrocortisol (5-alpha THF) and 5-beta tetrahydrocortisol (5-beta THF), reactions for which 5-alpha reductase and 5-beta-reductase are the rate-limiting factors, respectively. 5-Beta reductase is also the rate-limiting factor in the conversion of cortisone to tetrahydrocortisone.
Chemistry
Cortisol is a naturally occurring pregnane corticosteroid and is also known as 11β,17α,21-trihydroxypregn-4-ene-3,20-dione.
Animals
In animals, cortisol is often used as an indicator of stress and can be measured in blood,[90] saliva,[90] urine,[91] hair,[92] and faeces.[92][93]
See also
- Cortisone, a hormone
- Cortisol awakening response
- List of corticosteroids
- Membrane glucocorticoid receptor
References
- Scott E (22 September 2011). "Cortisol and Stress: How to Stay Healthy". About.com. Retrieved 29 November 2011.
- Taves MD, Gomez-Sanchez CE, Soma KK (July 2011). "Extra-adrenal glucocorticoids and mineralocorticoids: evidence for local synthesis, regulation, and function". American Journal of Physiology. Endocrinology and Metabolism. 301 (1): E11-24. doi:10.1152/ajpendo.00100.2011. PMC 3275156. PMID 21540450.
- Hoehn K, Marieb EN (2010). Human Anatomy & Physiology. San Francisco: Benjamin Cummings. ISBN 978-0-321-60261-9.
- Chyun YS, Kream BE, Raisz LG (February 1984). "Cortisol decreases bone formation by inhibiting periosteal cell proliferation". Endocrinology. 114 (2): 477–80. doi:10.1210/endo-114-2-477. PMID 6690287.
- Laycock JF (2013). Integrated endocrinology. Meeran, Karim. Chichester, West Sussex, UK: Wiley-Blackwell. ISBN 978-1-118-45064-2. OCLC 794973804.
- Martin PA, Crump MH (2003). "The adrenal gland". In Dooley MP, Pineda MH (eds.). McDonald's veterinary endocrinology and reproduction (5th ed.). Ames, Iowa: Iowa State Press. ISBN 978-0-8138-1106-2.
- Coderre L, Srivastava AK, Chiasson JL (June 1991). "Role of glucocorticoid in the regulation of glycogen metabolism in skeletal muscle". The American Journal of Physiology. 260 (6 Pt 1): E927–32. doi:10.1152/ajpendo.1991.260.6.E927. PMID 1905485.
- Macfarlane DP, Forbes S, Walker BR (May 2008). "Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome". The Journal of Endocrinology. 197 (2): 189–204. doi:10.1677/JOE-08-0054. PMID 18434349.
- Kuo T, McQueen A, Chen TC, Wang JC (2015). "Regulation of Glucose Homeostasis by Glucocorticoids". In Wang JC, Harris C (eds.). Glucocorticoid Signaling: From Molecules to Mice to Man. Advances in Experimental Medicine and Biology. Vol. 872. Springer. pp. 99–126. doi:10.1007/978-1-4939-2895-8_5. ISBN 978-1-4939-2895-8. PMC 6185996. PMID 26215992.
- Simmons PS, Miles JM, Gerich JE, Haymond MW (February 1984). "Increased proteolysis. An effect of increases in plasma cortisol within the physiologic range". The Journal of Clinical Investigation. 73 (2): 412–20. doi:10.1172/JCI111227. PMC 425032. PMID 6365973.
- Djurhuus CB, Gravholt CH, Nielsen S, Mengel A, Christiansen JS, Schmitz OE, Møller N (July 2002). "Effects of cortisol on lipolysis and regional interstitial glycerol levels in humans". American Journal of Physiology. Endocrinology and Metabolism. 283 (1): E172–7. doi:10.1152/ajpendo.00544.2001. PMID 12067858. S2CID 2609285.
- Elenkov IJ (June 2004). "Glucocorticoids and the Th1/Th2 balance". Annals of the New York Academy of Sciences. 1024 (1): 138–46. Bibcode:2004NYASA1024..138E. doi:10.1196/annals.1321.010. PMID 15265778. S2CID 9575617.
- Palacios R, Sugawara I (January 1982). "Hydrocortisone abrogates proliferation of T cells in autologous mixed lymphocyte reaction by rendering the interleukin-2 Producer T cells unresponsive to interleukin-1 and unable to synthesize the T-cell growth factor". Scandinavian Journal of Immunology. 15 (1): 25–31. doi:10.1111/j.1365-3083.1982.tb00618.x. PMID 6461917. S2CID 41292936.
- Besedovsky HO, Del Rey A, Sorkin E (1986). "Integration of Activated Immune Cell Products in Immune Endocrine Feedback Circuits". In Oppenheim JJ, Jacobs DM (eds.). Leukocytes and Host Defense. Progress in Leukocyte Biology. Vol. 5. New York: Alan R. Liss. p. 200.
- Demers Lawrence M (2008). "Adrenal Cortical Disorders". In Burtis Carl A, Ashwood Edward R, Bruns David E, Sawyer, Barbara G (eds.). Tietz Fundamentals of Clinical Chemistry. St. Louis, Missouri: Saunders El Sevier. p. 749-765.
- Murphy, Kenneth (2012). "The Humoral Immune Response". Janeway's Immunobiology, 8th ed. New York,NY: Garland Science Taylor & Francis Group. p. 387.
- Mavoungou E, Bouyou-Akotet MK, Kremsner PG (February 2005). "Effects of prolactin and cortisol on natural killer (NK) cell surface expression and function of human natural cytotoxicity receptors (NKp46, NKp44 and NKp30)". Clinical and Experimental Immunology. 139 (2): 287–96. doi:10.1111/j.1365-2249.2004.02686.x. PMC 1809301. PMID 15654827.
- Flohe L, Beckman R, Giertz H, Loschen G (1985). "Oxygen Centered Free Radicals as Mediators of Inflammation". In Sies H (ed.). Oxidative stress. London: Orlando. p. 405. ISBN 978-0-12-642760-8.
- Brown DF, Brown DD (2003). USMLE Step 1 Secrets: Questions You Will Be Asked on USMLE Step 1. Philadelphia: Hanley & Belfus. p. 63. ISBN 978-1-56053-570-6.
- King MB (2005). Lange Q & A. New York: McGraw-Hill, Medical Pub. Division. ISBN 978-0-07-144578-8.
- Baynes J, Dominiczak M (2009). Medical biochemistry. Mosby Elsevier. ISBN 978-0-323-05371-6.
- Knight RP, Kornfeld DS, Glaser GH, Bondy PK (February 1955). "Effects of intravenous hydrocortisone on electrolytes of serum and urine in man". The Journal of Clinical Endocrinology and Metabolism. 15 (2): 176–81. doi:10.1210/jcem-15-2-176. PMID 13233328.
- Deutsch E (April 1978). "[Pathogenesis of thrombocytopenia. 2. Distribution disorders, pseudo-thrombocytopenias]". Fortschritte der Medizin (in German). 96 (14): 761–2. PMID 346457.
- Kucharz EJ (1988). "Hormonal control of collagen metabolism. Part II". Endocrinologie. 26 (4): 229–37. PMID 3062759.
- Manchester, KL (1964). "Sites of Hormonal Regulation of Protein Metabolism". In Allison, NH; Munro JB (eds.). Mammalian Protein Metabolism. New York: Academic Press. p. 229? 273?.
- Husband AJ, Brandon MR, Lascelles AK (October 1973). "The effect of corticosteroid on absorption and endogenous production of immunoglobulins in calves". The Australian Journal of Experimental Biology and Medical Science. 51 (5): 707–10. doi:10.1038/icb.1973.67. PMID 4207041.
- Posey WC, Nelson HS, Branch B, Pearlman DS (December 1978). "The effects of acute corticosteroid therapy for asthma on serum immunoglobulin levels". The Journal of Allergy and Clinical Immunology. 62 (6): 340–8. doi:10.1016/0091-6749(78)90134-3. PMID 712020.
- McKay LI, Cidlowski JA (2003). "Physiologic and Pharmacologic Effects of Corticosteroids". In Kure DW, Pollock RE, Weichselbaum RR, Bast RC, Ganglier TS, Holland JF, Frei E (eds.). Holland-Frei Cancer Medicine (6th ed.). Hamilton, Ontario: Decker. ISBN 978-1-55009-213-4.
- Sandle GI, Keir MJ, Record CO (1981). "The effect of hydrocortisone on the transport of water, sodium, and glucose in the jejunum. Perfusion studies in normal subjects and patients with coeliac disease". Scandinavian Journal of Gastroenterology. 16 (5): 667–71. doi:10.3109/00365528109182028. PMID 7323700.
- Mason PA, Fraser R, Morton JJ, Semple PF, Wilson A (August 1977). "The effect of sodium deprivation and of angiotensin II infusion on the peripheral plasma concentrations of 18-hydroxycorticosterone, aldosterone and other corticosteroids in man". Journal of Steroid Biochemistry. 8 (8): 799–804. doi:10.1016/0022-4731(77)90086-3. PMID 592808.
- Gorbman A, Dickhoff WW, Vigna SR, Clark NB, Muller AF (1983). Comparative endocrinology. New York: Wiley. ISBN 978-0-471-06266-0.
- Muller AF, Oconnor CM (1958). An International Symposium on Aldosterone. Little Brown & Co. p. 58.
- Soffer LJ, Dorfman RI, Gabrilove JL (1961). The Human Adrenal Gland. Philadelphia: Lea & Febiger.
- Kokoshchuk GI, Pakhmurnyĭ BA (May 1979). "Role of glucocorticoids in regulating the acid-excreting function of the kidneys". Fiziologicheskii Zhurnal SSSR Imeni I. M. Sechenova. 65 (5): 751–4. PMID 110627.
- Kennedy R. "Cortisol (Hydrocortisone)". The Doctors' Medical Library. Archived from the original on 3 July 2013. Retrieved 14 June 2013.
- McAuley MT, Kenny RA, Kirkwood TB, Wilkinson DJ, Jones JJ, Miller VM (March 2009). "A mathematical model of aging-related and cortisol induced hippocampal dysfunction". BMC Neuroscience. 10: 26. doi:10.1186/1471-2202-10-26. PMC 2680862. PMID 19320982.
- Lundberg U (2010). "Neuroendocrine Measures". In Contrada R, Baum A (eds.). The Handbook of Stress Science: Biology, Psychology, and Health. New York: Springer Publishing Company. p. 351. ISBN 978-0-8261-1771-7. Retrieved 12 March 2020.
[...] epinephrine, norepinephrine, and cortisol are considered the most important 'stress hormones,' although a number of other hormones are also influenced by stress [...].
- Mescher EJ, Platzker AC, Ballard PL, Kitterman JA, Clements JA, Tooley WH (December 1975). "Ontogeny of tracheal fluid, pulmonary surfactant, and plasma corticoids in the fetal lamb". Journal of Applied Physiology. 39 (6): 1017–21. doi:10.1152/jappl.1975.39.6.1017. PMID 2573.
- Hennessy DP, Coghlan JP, Hardy KJ, Scoggins BA, Wintour EM (October 1982). "The origin of cortisol in the blood of fetal sheep". The Journal of Endocrinology. 95 (1): 71–9. doi:10.1677/joe.0.0950071. PMID 7130892.
- Magyar DM, Fridshal D, Elsner CW, Glatz T, Eliot J, Klein AH, Lowe KC, Buster JE, Nathanielsz PW (July 1980). "Time-trend analysis of plasma cortisol concentrations in the fetal sheep in relation to parturition". Endocrinology. 107 (1): 155–9. doi:10.1210/endo-107-1-155. PMID 7379742.
- Ricketts AP, Flint AP (August 1980). "Onset of synthesis of progesterone by ovine placenta". The Journal of Endocrinology. 86 (2): 337–47. doi:10.1677/joe.0.0860337. PMID 6933207.
- Al-Gubory KH, Solari A, Mirman B (1999). "Effects of luteectomy on the maintenance of pregnancy, circulating progesterone concentrations and lambing performance in sheep". Reproduction, Fertility, and Development. 11 (6): 317–22. doi:10.1071/RD99079. PMID 10972299.
- Mustoe AC, Birnie AK, Korgan AC, Santo JB, French JA (February 2012). "Natural variation in gestational cortisol is associated with patterns of growth in marmoset monkeys (Callithrix geoffroyi)". General and Comparative Endocrinology. 175 (3): 519–26. doi:10.1016/j.ygcen.2011.12.020. PMC 3268124. PMID 22212825.
- "You & Your Hormones: Cortisol". the Society for Endocrinology (Last updated). 24 October 2013. Archived from the original on 21 October 2014. Retrieved 24 November 2014.
- Hanukoglu A, Fried D, Nakash I, Hanukoglu I (November 1995). "Selective increases in adrenal steroidogenic capacity during acute respiratory disease in infants". Eur J Endocrinol. 133 (5): 552–6. doi:10.1530/eje.0.1330552. PMID 7581984. S2CID 44439040.
- Biochemistry Reference Ranges at Good Hope Hospital Retrieved 8 November 2009
- Derived from molar values using molar mass of 362 g/mol
- Converted from µg/24h, using molar mass of 362.460 g/mol
- Görges R, Knappe G, Gerl H, Ventz M, Stahl F (April 1999). "Diagnosis of Cushing's syndrome: re-evaluation of midnight plasma cortisol vs urinary free cortisol and low-dose dexamethasone suppression test in a large patient group". Journal of Endocrinological Investigation. 22 (4): 241–9. doi:10.1007/bf03343551. PMID 10342356. S2CID 1239611.
- MedlinePlus Encyclopedia: Cortisol – urine
- Converted from nmol/24h, using molar mass of 362.460 g/mol
- Izawa S, Sugaya N, Ogawa N, Shirotsuki K, Nomura S (April 2021). "A validation study on fingernail cortisol: correlations with one-month cortisol levels estimated by hair and saliva samples". Stress (Amsterdam, Netherlands). 24 (6): 734–741. doi:10.1080/10253890.2021.1895113. PMID 33792492. S2CID 232481968.
- Turpeinen U, Hämäläinen E (December 2013). "Determination of cortisol in serum, saliva and urine". Best Practice & Research. Clinical Endocrinology & Metabolism. 27 (6): 795–801. doi:10.1016/j.beem.2013.10.008. PMID 24275191.
- Dolomie-Fagour L, Corcuff JB (2008). "[Is free plasmatic cortisol measurement useful in intensive care unit?]". Annales de Biologie Clinique (in French). 66 (1): 31–41. doi:10.1684/abc.2008.0189 (inactive 31 July 2022). PMID 18227002.
{{cite journal}}: CS1 maint: DOI inactive as of July 2022 (link) - Maidana P, Bruno OD, Mesch V (2013). "[A critical analysis of cortisol measurements: an update]". Medicina (in Spanish). 73 (6): 579–84. PMID 24356273.
- Verbeeten KC, Ahmet AH (January 2018). "The role of corticosteroid-binding globulin in the evaluation of adrenal insufficiency". Journal of Pediatric Endocrinology & Metabolism. 31 (2): 107–115. doi:10.1515/jpem-2017-0270. PMID 29194043. S2CID 28588420.
- El-Farhan N, Rees DA, Evans C (May 2017). "Measuring cortisol in serum, urine and saliva - are our assays good enough?". Annals of Clinical Biochemistry. 54 (3): 308–322. doi:10.1177/0004563216687335. PMID 28068807. S2CID 206397561.
- "Cushing's Syndrome". The Lecturio Medical Concept Library. Retrieved 11 July 2021.
- "Cushing's Syndrome". National Endocrine and Metabolic Diseases Information Service (NEMDIS). July 2008. Archived from the original on 10 February 2015. Retrieved 16 March 2015.
These benign, or noncancerous, tumors of the pituitary gland secrete extra ACTH. Most people with the disorder have a single adenoma. This form of the syndrome, known as Cushing's disease
- Forbis P (2005). Stedman's medical eponyms (2nd ed.). Baltimore, Md.: Lippincott Williams & Wilkins. p. 167. ISBN 978-0-7817-5443-9.
- Davies E, Kenyon CJ, Fraser R (June 1985). "The role of calcium ions in the mechanism of ACTH stimulation of cortisol synthesis". Steroids. 45 (6): 551–60. doi:10.1016/0039-128X(85)90019-4. PMID 3012830. S2CID 24454836.
- Plotsky PM, Otto S, Sapolsky RM (September 1986). "Inhibition of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation by delayed glucocorticoid feedback". Endocrinology. 119 (3): 1126–30. doi:10.1210/endo-119-3-1126. PMID 3015567.
- Minton JE, Parsons KM (March 1993). "Adrenocorticotropic hormone and cortisol response to corticotropin-releasing factor and lysine vasopressin in pigs". Journal of Animal Science. 71 (3): 724–9. doi:10.2527/1993.713724x. PMID 8385088.
- Fairchild SS, Shannon K, Kwan E, Mishell RI (February 1984). "T cell-derived glucosteroid response-modifying factor (GRMFT): a unique lymphokine made by normal T lymphocytes and a T cell hybridoma". Journal of Immunology. 132 (2): 821–7. PMID 6228602.
- Dvorak M (1971). "Plasma 17-Hydroxycorticosteroid Levels in Healthy and Diarrheic Calves". British Veterinarian Journal. 127: 372.
- Stith RD, McCallum RE (1986). "General effect of endotoxin on glucocorticoid receptors in mammalian tissues". Circulatory Shock. 18 (4): 301–9. PMID 3084123.
- Mikosha AS, Pushkarov IS, Chelnakova IS, Remennikov GY (1991). "Potassium Aided Regulation of Hormone Biosynthesis in Adrenals of Guinea Pigs Under Action of Dihydropyridines: Possible Mechanisms of Changes in Steroidogenesis Induced by 1,4, Dihydropyridines in Dispersed Adrenocorticytes". Fiziol. [Kiev]. 37: 60.
- "Ameer Saadallah Al – Zacko" (PDF). Archived from the original (PDF) on 11 November 2013. Retrieved 11 July 2013.
- Mendelsohn FA, Mackie C (July 1975). "Relation of intracellular K+ and steroidogenesis in isolated adrenal zona glomerulosa and fasciculata cells". Clinical Science and Molecular Medicine. 49 (1): 13–26. doi:10.1042/cs0490013. PMID 168026. S2CID 24873537.
- Ueda Y, Honda M, Tsuchiya M, Watanabe H, Izumi Y, Shiratsuchi T, Inoue T, Hatano M (April 1982). "Response of plasma ACTH and adrenocortical hormones to potassium loading in essential hypertension". Japanese Circulation Journal. 46 (4): 317–22. doi:10.1253/jcj.46.317. PMID 6283190.
- Bauman K, Muller J (1972). "Effect of potassium on the final status of aldosterone biosynthesis in the rat. I 18-hydroxylation and 18hydroxy dehydrogenation. II beta-hydroxylation". Acta Endocrinol. 69 (4): I 701–717, II 718–730. doi:10.1530/acta.0.0690701. PMID 5067076.
- LaCelle PL, Morgan ES, Atwater EC (1964). "An investigation of total body potassium in patients with rheumatoid arthritis". Proceedings of the Annual Meeting of the American Rheumatism Association, Arthritis and Rheumatism. 7 (3): 321.
- Brody S, Preut R, Schommer K, Schürmeyer TH (January 2002). "A randomized controlled trial of high dose ascorbic acid for reduction of blood pressure, cortisol, and subjective responses to psychological stress". Psychopharmacology. 159 (3): 319–24. doi:10.1007/s00213-001-0929-6. PMID 11862365. S2CID 2778669.
- Silverman MN, Pearce BD, Biron CA, Miller AH (2005). "Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection". Viral Immunology. 18 (1): 41–78. doi:10.1089/vim.2005.18.41. PMC 1224723. PMID 15802953.
- Robson PJ, Blannin AK, Walsh NP, Castell LM, Gleeson M (February 1999). "Effects of exercise intensity, duration and recovery on in vitro neutrophil function in male athletes". International Journal of Sports Medicine. 20 (2): 128–35. doi:10.1055/s-2007-971106. PMID 10190775.
- Fuqua JS, Rogol AD (July 2013). "Neuroendocrine alterations in the exercising human: implications for energy homeostasis". Metabolism. 62 (7): 911–21. doi:10.1016/j.metabol.2013.01.016. PMID 23415825.
Cortisol has wide-ranging effects, including alterations of carbohydrate, protein, and lipid metabolism; catabolic effects on skin, muscle, connective tissue, and bone; immunomodulatory effects; blood pressure and circulatory system regulation; and effects on mood and central nervous system function. In the short term, activation of the HPA axis in response to stress is adaptive. However, long-term stress promoting chronic exposure of tissues to high cortisol concentrations becomes maladaptive. ... Exercise, particularly sustained aerobic activity, is a potent stimulus of cortisol secretion. The circulating concentrations of cortisol are directly proportional to the intensity of exercise as measured by oxygen uptake. As is the case for the GH/IGF-1 and HPG axes, the HPA axis also receives many other inputs, including the light/dark cycle, feeding schedules, immune regulation, and many neurotransmitters that mediate the effects of exercise and physical and psychic stress [52]. ... The HPA is activated by stress, whether physical (exercise) or psychological. Increased cortisol production, along with activation of the sympathetic nervous system, affects whole-body metabolism. This is apparently part of the catabolic response of the entire organism, with the purpose of mobilizing metabolic fuels that are subsequently broken down to produce energy and to dampen the threat or perceived threat. ... Thus, a negative net energy balance leads to activation of the HPA axis and the circulating concomitants of the catabolic state in an attempt to keep core processes functional, realizing that the stress of exercise has no effect on cortisol and circulating metabolic substrates beyond the impact of the exercise energy expenditure on energy availability [60]. Thuma et al. [61] had already made the important observation that the reported differences in cortisol levels pre- and postexercise depended on whether this difference was measured from a single pretest level or from the physiologic circadian baseline as determined in an independent session in the resting state. By this analytical technique, these investigators showed that increasing energy expenditure led to significant cortisol release. This release was apparent if they subtracted the physiologic circadian baseline from the postexercise value.
- Smith JL, Gropper SA, Groff JL (2009). Advanced nutrition and humanmetabolism. Belmont, CA: Wadsworth Cengage Learning. p. 247. ISBN 978-0-495-11657-8.
- Whittaker J, Harris M (March 2022). "Low-carbohydrate diets and men's cortisol and testosterone: Systematic review and meta-analysis". Nutrition and Health: 2601060221083079. doi:10.1177/02601060221083079. PMID 35254136. S2CID 247251547.
- Häggström M, Richfield D (2014). "Diagram of the pathways of human steroidogenesis". WikiJournal of Medicine. 1 (1). doi:10.15347/wjm/2014.005. ISSN 2002-4436.
- Willett LB, Erb RE (January 1972). "Short term changes in plasma corticoids in dairy cattle". Journal of Animal Science. 34 (1): 103–11. doi:10.2527/jas1972.341103x. PMID 5062063.
- Margioris AN, Tsatsanis C (2011). "ACTH Action on the Adrenal". In Chrousos G (ed.). Adrenal physiology and diseases. Endotext.org. Archived from the original on 29 November 2011. Retrieved 5 June 2012.
- Finken MJ, Andrews RC, Andrew R, Walker BR (September 1999). "Cortisol metabolism in healthy young adults: sexual dimorphism in activities of A-ring reductases, but not 11beta-hydroxysteroid dehydrogenases". The Journal of Clinical Endocrinology and Metabolism. 84 (9): 3316–3321. doi:10.1210/jcem.84.9.6009. PMID 10487705.
- Dammann C, Stapelfeld C, Maser E (April 2019). "Expression and activity of the cortisol-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 is tissue and species-specific". Chemico-Biological Interactions. 303: 57–61. doi:10.1016/j.cbi.2019.02.018. PMID 30796905.
- Atanasov AG, Nashev LG, Schweizer RA, Frick C, Odermatt A (July 2004). "Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase". FEBS Letters. 571 (1–3): 129–133. doi:10.1016/j.febslet.2004.06.065. PMID 15280030.
- Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM (October 2004). "11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response". Endocrine Reviews. 25 (5): 831–66. doi:10.1210/er.2003-0031. PMID 15466942.
- "6beta-Hydroxycortisol".
- Luceri F, Fattori S, Luceri C, Zorn M, Mannaioni P, Messeri G (December 2001). "Gas chromatography-mass spectrometry measurement of 6beta-OH-cortisol/cortisol ratio in human urine: a specific marker of enzymatic induction". Clin Chem Lab Med. 39 (12): 1234–9. doi:10.1515/CCLM.2001.198. PMID 11798083. S2CID 12216877.
- Huang FR, Zhou C, Zhang XY, Shen Y, Zhang HW, Wang YQ, Sun LN (October 2021). "Impact of CYP2C19 genotype on voriconazole exposure and effect of voriconazole on the activity of CYP3A in patients with haematological malignancies". Xenobiotica. 51 (10): 1199–1206. doi:10.1080/00498254.2021.1969481. PMID 34402388. S2CID 237150260.
- Aquinos BM, García Arabehety J, Canteros TM, de Miguel V, Scibona P, Fainstein-Day P (2021). "[Adrenal crisis associated with modafinil use]". Medicina (B Aires) (in Spanish). 81 (5): 846–849. PMID 34633961.
- van Staaveren N, Teixeira DL, Hanlon A, Boyle LA (2015). "The effect of mixing entire male pigs prior to transport to slaughter on behaviour, welfare and carcass lesions". PLOS ONE. 10 (4): e0122841. Bibcode:2015PLoSO..1022841V. doi:10.1371/journal.pone.0122841. PMC 4382277. PMID 25830336.
- Schalke E, Stichnoth J, Ott S, Jones-Baade R (2007). "Clinical signs caused by the use of electric training collars on dogs in everyday life situations". Applied Animal Behaviour Science. 105 (4): 369–380. doi:10.1016/j.applanim.2006.11.002.
- Accorsi PA, Carloni E, Valsecchi P, Viggiani R, Gamberoni M, Tamanini C, Seren E (January 2008). "Cortisol determination in hair and faeces from domestic cats and dogs". General and Comparative Endocrinology. 155 (2): 398–402. doi:10.1016/j.ygcen.2007.07.002. PMID 17727851.
- Möstl E, Messmann S, Bagu E, Robia C, Palme R (December 1999). "Measurement of glucocorticoid metabolite concentrations in faeces of domestic livestock". Zentralblatt für Veterinarmedizin. Reihe A. 46 (10): 621–631. doi:10.1046/j.1439-0442.1999.00256.x. PMID 10638300.
External links
- Cortisol MS Spectrum
- Cortisol: analyte monograph – The Association for Clinical Biochemistry and Laboratory Medicine