Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is a condition in which the heart becomes enlarged and cannot pump blood effectively.[3] Symptoms vary from none to feeling tired, leg swelling, and shortness of breath.[2] It may also result in chest pain or fainting.[2] Complications can include heart failure, heart valve disease, or an irregular heartbeat.[3][4]
| Dilated cardiomyopathy | |
|---|---|
| Other names | Congestive cardiomyopathy, idiopathic cardiomyopathy, primary cardiomyopathy[1] |
 | |
| Mouse heart slice showing dilated cardiomyopathy | |
| Specialty | Cardiology |
| Symptoms | Feeling tired, leg swelling, shortness of breath, chest pain, fainting[2] |
| Complications | Heart failure, heart valve disease, irregular heartbeat[3][4] |
| Usual onset | Middle age[5] |
| Types | Tachycardia-induced,[6][7] others |
| Causes | Genetics, alcohol, cocaine, certain toxins, complications of pregnancy, in many cases the cause remains unclear, certain infections[8][9][7] |
| Diagnostic method | Supported by electrocardiogram, chest X-ray, echocardiogram[9] |
| Differential diagnosis | Coronary artery disease, heart valve disease, pulmonary embolism, other cardiomyopathy[5] |
| Treatment | Lifestyle changes, medications, implantable cardioverter defibrillator, cardiac resynchronization therapy (CRT), heart transplant[9] |
| Medication | ACE inhibitor, beta blocker, diuretic, blood thinners[9] |
| Prognosis | Five-year survival rate ~50%[9] |
| Frequency | 1 in 2500[9] |
Causes include genetics, alcohol, cocaine, certain toxins, complications of pregnancy, and certain infections.[8][9] Coronary artery disease and high blood pressure may play a role, but are not the primary cause.[5][8] In many cases the cause remains unclear.[8] It is a type of cardiomyopathy, a group of diseases that primarily affects the heart muscle.[3] The diagnosis may be supported by an electrocardiogram, chest X-ray, or echocardiogram.[9]
In those with heart failure, treatment may include medications in the ACE inhibitor, beta blocker, and diuretic families.[9] A low salt diet may also be helpful.[5] In those with certain types of irregular heartbeat, blood thinners or an implantable cardioverter defibrillator may be recommended. Cardiac resynchronization therapy (CRT) may be necessary.[9] If other measures are not effective a heart transplant may be an option in some.[9]
About 1 per 2,500 people is affected.[9] It occurs more frequently in men than women.[10] Onset is most often in middle age.[5] Five-year survival rate is about 50%.[9] It can also occur in children and is the most common type of cardiomyopathy in this age group.[9]
Signs and symptoms
Dilated cardiomyopathy develops insidiously, and may not initially cause symptoms significant enough to impact on quality of life.[11][12] Nevertheless, many people experience significant symptoms. These might include:[13]
- Shortness of breath
- Syncope (fainting)
- Angina, but only in the presence of ischemic heart disease
A person who has dilated cardiomyopathy may have an enlarged heart, with pulmonary edema and an elevated jugular venous pressure and a low pulse pressure. Signs of mitral and tricuspid regurgitation may be present.[12]
Causes
Although in many cases no cause is apparent, dilated cardiomyopathy is probably the result of damage to the myocardium produced by a variety of toxic, metabolic, or infectious agents. In many cases the cause remains unclear. It may be due to fibrous change of the myocardium from a previous myocardial infarction. Or, it may be the late sequelae of acute viral myocarditis, such as with Coxsackie B virus and other enteroviruses[14] possibly mediated through an immunologic mechanism.[15] Specific autoantibodies are detectable in some cases.[16]
Other causes include:
- Chagas disease, due to Trypanosoma cruzi. This is the most common infectious cause of dilated cardiomyopathy in Latin America[17]
- Pregnancy: Dilated cardiomyopathy occurs late in gestation or several weeks to months postpartum as a peripartum cardiomyopathy.[14] It is reversible in half of cases.[14]
- Alcohol use disorder (alcoholic cardiomyopathy)[14]
- Non-alcoholic toxic insults include administration of certain chemotherapeutic agents, in particular doxorubicin (Adriamycin), and cobalt.[14]
- Thyroid disease[12]
- Inflammatory diseases such as sarcoidosis and connective tissue diseases[12]
- Tachycardia-induced cardiomyopathy[7]
- Muscular dystrophy
- Tuberculosis: 1 to 2% of TB cases.[18]
- Autoimmune mechanisms[19]
- Thiamine deficiency
Recent studies have shown that those subjects with an extremely high occurrence (several thousands a day) of premature ventricular contractions (extrasystole) can develop dilated cardiomyopathy. In these cases, if the extrasystole are reduced or removed (for example, via ablation therapy) the cardiomyopathy usually regresses.[20][21]
Genetics
| Genetic associations with dilated cardiomyopathy | |||
|---|---|---|---|
| Type | OMIM | Gene | Locus |
| CMD1A | 115200 | LMNA | 1q21 |
| CMD1B | 600884 | unknown (TMOD1 candidate) | 9q13 |
| CMD1C | 601493 | LDB3 | 10q22-q23 |
| CMD1D | 601494 | TNNT2 | 1q32 |
| CMD1E | 601154 | SCN5A | 3p |
| CMD1F | 602067 | 6q23 | |
| CMD1G | 604145 | TTN | 2q31 |
| CMD1H | 604288 | 2q14-q22 | |
| CMD1I | 604765 | DES | |
| CMD1K | 605582 | 6q12-q16 | |
| CMD1L | 606685 | SGCD | 5q33 |
| CMD1M | 607482 | CSRP3 | 11p15.1 |
| CMD1N | 607487 | TCAP | 17q12 |
| CMD1O | 608569 | ABCC9 | 12p12.1 |
| CMD1P | 609909 | PLN | 6q22.1 |
| CMD1Q | 609915 | 7q22.3-q31.1 | |
| CMD1R | ACTC | 15q14 | |
| CMD1S | MYH7 | 14q12 | |
| CMD1T | TMPO | 12q22 | |
| CMD1U | PSEN1 | 14q24.3 | |
| CMD1V | PSEN2 | 1q31-q42 | |
| CMD1W | 611407 | VCL | 10q22-q23 |
| CMD1X | FCMD | 9q31 | |
| CMD1Y | 611878 | TPM1 | 15q22.1 |
| CMD1Z | 611879 | TNNC1 | 3p21.3-p14.3 |
| CMD1AA | 612158 | ACTN2 | 1q42-q43 |
| CMD2A | 611880 | TNNI3 | 19q13.4 |
| CMD3A | 300069 | TAZ | Xq28 |
| CMD3B | 302045 | DMD | Xp21.2 |
| ALPK3 | 15q25.3 | ||
About 25–35% of affected individuals have familial forms of the disease,[14] with most mutations affecting genes encoding cytoskeletal proteins,[14] while some affect other proteins involved in contraction.[22] The disease is genetically heterogeneous, but the most common form of its transmission is an autosomal dominant pattern.[14] Autosomal recessive (as found, for example, in Alström syndrome[14]), X-linked (as in Duchenne muscular dystrophy), and mitochondrial inheritance of the disease is also found.[23] Some relatives of those affected by dilated cardiomyopathy have preclinical, asymptomatic heart-muscle changes.[24]
Other cytoskeletal proteins involved in DCM include α-cardiac actin, desmin, and the nuclear lamins A and C.[14] Mitochondrial deletions and mutations presumably cause DCM by altering myocardial ATP generation.[14]
Kayvanpour et al. performed 2016 a meta-analysis with the largest dataset available on genotype-phenotype associations in DCM and mutations in lamin (LMNA), phospholamban (PLN), RNA Binding Motif Protein 20 (RBM20), Cardiac Myosin Binding Protein C (MYBPC3), Myosin Heavy Chain 7 (MYH7), Cardiac Troponin T 2 (TNNT2), and Cardiac Troponin I (TNNI3). They also reviewed recent studies investigating genotype-phenotype associations in DCM patients with titin (TTN) mutations. LMNA and PLN mutation carriers showed a high prevalence of cardiac transplantation and ventricular arrhythmia. Dysrhythmias and sudden cardiac death (SCD) was shown to occur even before the manifestation of DCM and heart failure symptoms in LMNA mutation carriers.[25]
Pathophysiology
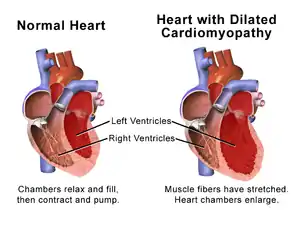
The progression of heart failure is associated with left ventricular remodeling, which manifests as gradual increases in left ventricular end-diastolic and end-systolic volumes, wall thinning, and a change in chamber geometry to a more spherical, less elongated shape. This process is usually associated with a continuous decline in ejection fraction. The concept of cardiac remodeling was initially developed to describe changes that occur in the days and months following myocardial infarction.[26]
Compensation effects
As DCM progresses, two compensatory mechanisms are activated in response to impaired myocyte contractility and reduced stroke volume:[12]
- Frank-Starling law
- Neurohormonal feedback, via activation of the sympathetic nervous system and the renin-angiotensin system.
These responses initially compensate for decreased cardiac output and maintain those with DCM as asymptomatic. Eventually, however, these mechanisms become detrimental, intravascular volume becomes too great, and progressive dilatation leads to heart failure symptoms.
Computational models
Cardiac dilatation is a transversely isotropic, irreversible process resulting from excess strains on the myocardium.[27] A computation model of volumetric, isotropic, and cardiac wall growth predicts the relationship between cardiac strains (e.g. volume overload after myocardial infarction) and dilation using the following governing equations:

where is elastic volume stretch that is reversible and is irreversible, isotropic volume growth described by:



where is a vector, which points along a cardiomyocyte's long axis and is the cardiomyocyte stretch due to growth. The total cardiomyocyte growth is given by:



The above model reveals a gradual dilation of the myocardium, especially the ventricular myocardium, to support the blood volume overload in the chambers. Dilation manifests itself in an increase in total cardiac mass and cardiac diameter. Cardiomyocytes reach their maximum length of 150 m in the endocardium and 130 m in the epicardium by the addition of sarcomeres.[27] Due to the increase in diameter, the dilated heart appears spherical in shape, as opposed the elliptical shape of a healthy human heart. In addition, the ventricular walls maintain the same thickness, characteristic of pathophysiological cardiac dilation.

Valvular effects
As the ventricles enlarge, both the mitral and tricuspid valves may lose their ability to come together properly. This loss of coaptation may lead to mitral and tricuspid regurgitation. As a result, those with DCM are at increased risk of atrial fibrillation. Furthermore, stroke volume is decreased and a greater volume load is placed on the ventricle, thus increasing heart failure symptoms.[12]
Diagnosis
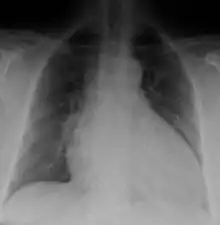
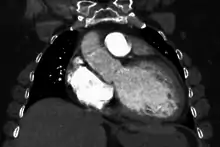
Generalized enlargement of the heart is seen upon normal chest X-ray. Pleural effusion may also be noticed, which is due to pulmonary venous hypertension.[28]
The electrocardiogram often shows sinus tachycardia or atrial fibrillation, ventricular arrhythmias, left atrial enlargement, and sometimes intraventricular conduction defects and low voltage. When left bundle-branch block (LBBB) is accompanied by right axis deviation (RAD), the rare combination is considered to be highly suggestive of dilated or congestive cardiomyopathy.[29][30] Echocardiogram shows left ventricular dilatation with normal or thinned walls and reduced ejection fraction. Cardiac catheterization and coronary angiography are often performed to exclude ischemic heart disease.[28]
Genetic testing can be important, since one study has shown that gene mutations in the TTN gene (which codes for a protein called titin) are responsible for "approximately 25% of familial cases of idiopathic dilated cardiomyopathy and 18% of sporadic cases."[31] The results of the genetic testing can help the doctors and patients understand the underlying cause of the dilated cardiomyopathy. Genetic test results can also help guide decisions on whether a patient's relatives should undergo genetic testing (to see if they have the same genetic mutation) and cardiac testing to screen for early findings of dilated cardiomyopathy.[28]
Cardiac magnetic resonance imaging (cardiac MRI) may also provide helpful diagnostic information in patients with dilated cardiomyopathy.[32]
Treatment
Medical therapy
Drug therapy can slow down progression and in some cases even improve the heart condition. Standard therapy may include salt restriction, ACE inhibitors, diuretics, and beta blockers.[12] Anticoagulants may also be used for antithrombotic therapy. There is some evidence for the benefits of coenzyme Q10 in treating heart failure.[33][34][35]
Electrical treatment
Artificial pacemakers may be used in patients with intraventricular conduction delay, and implantable cardioverter-defibrillators in those at risk of arrhythmia. These forms of treatment have been shown to prevent sudden cardiac death, improve symptoms, and reduce hospitalization in patients with systolic heart failure.[36] In addition, an implantable cardioverter-defibrillator should be considered as a therapeutic option for the primary prevention of sudden cardiac death in patients with a confirmed LMNA mutation responsible for dilated cardiomyopathy disease phenotype and clinical risk factors.[37] A novel risk score calculator has been developed that allows calculation of risk of sustained ventricular arrhythmia in the next 5 years in patients with DCM [38] https://www.ikard.pl/SVA/
Surgical treatment
In patients with advanced disease who are refractory to medical therapy, heart transplantation may be considered. For these people, 1-year survival approaches 90% and over 50% survive greater than 20 years.[36]
Epidemiology
Although the disease is more common in African-Americans than in Caucasians,[39] it may occur in any patient population.
Research directions
Therapies that support reverse remodeling have been investigated, and this may suggests a new approach to the prognosis of cardiomyopathies (see ventricular remodeling).[26][40]
Animals
In some types of animals, both a hereditary and acquired version of dilated cardiomyopathy has been documented.
Dogs
Dilated cardiomyopathy is a heritable disease in some dog breeds, including the Boxer, Dobermann, Great Dane, Irish Wolfhound, and St Bernard.[41] Treatment is based on medication, including ACE inhibitors, loop diuretics, and phosphodiesterase inhibitors.
An acquired variation of dilated cardiomyopathy describing a link between certain diets was discovered in 2019 by researchers at University of California, Davis School of Veterinary Medicine who published a report on the development of dilated cardiomyopathy in dog breeds lacking the genetic predisposition, particularly in Golden Retrievers.[42] The diets associated with DCM were described as "BEG" (boutique, exotic-ingredient, and/or grain-free) dog foods,[43] as well as legume-rich diets.[44] For treating diet-related DCM, food changes, taurine and carnitine supplementation may be indicated even if the dog does not have a documented taurine or carnitine deficiency although the cost of carnitine supplementation may be viewed as prohibitive by some[45]
Cats
Dilated cardiomyopathy is also a disease affecting some cat breeds, including the Oriental Shorthair, Burmese, Persian, and Abyssinian. In cats, taurine deficiency is the most common cause of dilated cardiomyopathy.[46] As opposed to these hereditary forms, non-hereditary DCM used to be common in the overall cat population before the addition of taurine to commercial cat food.
Other animals
There is also a high incidence of heritable dilated cardiomyopathy in captive Golden Hamsters (Mesocricetus auratus), due in no small part to their being highly inbred. The incidence is high enough that several strains of Golden Hamster have been developed to serve as animal models in clinical testing for human forms of the disease.[47]
References
- "Other Names for Cardiomyopathy". NHLBI. June 22, 2016. Retrieved 31 August 2016.
- "What Are the Signs and Symptoms of Cardiomyopathy?". NHLBI. 22 June 2016. Retrieved 10 November 2017.
- "What Is Cardiomyopathy?". NHLBI. 22 June 2016. Retrieved 10 November 2017.
- "Types of Cardiomyopathy". NHLBI. 22 June 2016. Retrieved 10 November 2017.
- Ferri FF (2017). Ferri's Clinical Advisor 2018 E-Book: 5 Books in 1. Elsevier Health Sciences. p. 244. ISBN 9780323529570.
- Pérez-Silva A, Merino JL (March 2009). "Tachycardia-induced cardiomyopathy". e-Journal of Cardiology Practice. 17: 16.
Tachycardia-induced cardiomyopathy is a reversible cause of heart failure and dilated cardiomyopathy. Tachycardia-induced cardiomyopathy should be considered in all patients with a dilated cardiomyopathy of uncertain origin and who have tachycardia or atrial fibrillation with a fast ventricular rate.
- Umana E, Solares CA, Alpert MA (January 2003). "Tachycardia-induced cardiomyopathy". The American Journal of Medicine. 114 (1): 51–5. doi:10.1016/S0002-9343(02)01472-9. PMID 12543289.
- "What Causes Cardiomyopathy?". NHLBI. 22 June 2016. Retrieved 10 November 2017.
- Weintraub RG, Semsarian C, Macdonald P (July 2017). "Dilated cardiomyopathy". Lancet. 390 (10092): 400–414. doi:10.1016/S0140-6736(16)31713-5. PMID 28190577. S2CID 46801202.
- "Who Is at Risk for Cardiomyopathy? - NHLBI, NIH". NHLBI. 22 June 2016. Retrieved 10 November 2017.
- Watkins H, Ashrafian H, Redwood C (April 2011). "Inherited cardiomyopathies". The New England Journal of Medicine. 364 (17): 1643–56. doi:10.1056/nejmra0902923. PMID 21524215.
- Pathophysiology of heart disease : a collaborative project of medical students and faculty. Lilly, Leonard S., Harvard Medical School. (5th ed.). Baltimore, MD: Wolters Kluwer/Lippincott Williams & Wilkins. 2011. ISBN 9781605477237. OCLC 649701807.
{{cite book}}: CS1 maint: others (link) - "Dilated Cardiomyopathy". The Lecturio Medical Concept Library. Retrieved 11 July 2021.
- Mitchell RS, Kumar V, Abbas AK, Fausto N (2007). Robbins Basic Pathology (8th ed.). Philadelphia: Saunders. ISBN 978-1-4160-2973-1.
- Martino TA, Liu P, Sole MJ (February 1994). "Viral infection and the pathogenesis of dilated cardiomyopathy". Circulation Research. 74 (2): 182–8. doi:10.1161/01.res.74.2.182. PMID 8293557.
- Yoshikawa T, Baba A, Nagatomo Y. Autoimmune mechanisms underlying dilated cardiomyopathy. Circ J. 2009 Apr;73(4):602-7. doi: 10.1253/circj.cj-08-1151. Epub 2009 Feb 26. PMID 19246813.
- Suboc T (October 2019). "Dilated Cardiomyopathy - Cardiovascular Disorders". Merck Manual. Merck & Co., Inc.
- Agarwal R, Malhotra P, Awasthi A, Kakkar N, Gupta D (April 2005). "Tuberculous dilated cardiomyopathy: an under-recognized entity?". BMC Infectious Diseases. 5: 29. doi:10.1186/1471-2334-5-29. PMC 1090580. PMID 15857515.
- San Martín MA, García A, Rodríguez FJ, Terol I (May 2002). "[Dilated cardiomyopathy and autoimmunity: an overview of current knowledge and perspectives]". Revista Espanola de Cardiologia (in Spanish). 55 (5): 514–24. doi:10.1016/s0300-8932(02)76644-x. PMID 12015932. S2CID 73293322. Archived from the original on 2009-01-09.
- Belhassen B (April 2005). "Radiofrequency ablation of "benign" right ventricular outflow tract extrasystoles: a therapy that has found its disease?". Journal of the American College of Cardiology. 45 (8): 1266–8. doi:10.1016/j.jacc.2005.01.028. PMID 15837260.
- Shiraishi H, Ishibashi K, Urao N, Tsukamoto M, Hyogo M, Keira N, et al. (November 2002). "A case of cardiomyopathy induced by premature ventricular complexes". Circulation Journal. 66 (11): 1065–7. doi:10.1253/circj.66.1065. PMID 12419942.
- Ross J (March 2002). "Dilated cardiomyopathy: concepts derived from gene deficient and transgenic animal models". Circulation Journal. 66 (3): 219–24. doi:10.1253/circj.66.219. PMID 11922267.
- Schönberger J, Seidman CE (August 2001). "Many roads lead to a broken heart: the genetics of dilated cardiomyopathy". American Journal of Human Genetics. 69 (2): 249–60. doi:10.1086/321978. PMC 1235300. PMID 11443548.
- Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ (July 2005). "Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease". Annals of Internal Medicine. 143 (2): 108–15. doi:10.7326/0003-4819-143-2-200507190-00009. PMID 16027452. S2CID 22278646.
- Kayvanpour E, Sedaghat-Hamedani F, Amr A, Lai A, Haas J, Holzer DB, et al. (February 2017). "Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals". Clinical Research in Cardiology. 106 (2): 127–139. doi:10.1007/s00392-016-1033-6. PMID 27576561. S2CID 27511518.
- Pieske B (2004). "Reverse remodeling in heart failure – fact or fiction?". Eur Heart J Suppl. 6: D66–78. doi:10.1016/j.ehjsup.2004.05.019.
- Goektepe S, Abilez OJ, Kuhl E (2010). "Generic approach towards finite growth with examples of athlete's heart, cardiac dilation, and cardiac wall thickening". Mechanics and Physics of Solids. 58 (10): 1661–1680. Bibcode:2010JMPSo..58.1661G. doi:10.1016/j.jmps.2010.07.003.
- "Dilated Cardiomyopathy". The Lecturio Medical Concept Library. Retrieved 25 August 2021.
- Nikolic G, Marriott HJ (October 1985). "Left bundle branch block with right axis deviation: a marker of congestive cardiomyopathy". Journal of Electrocardiology. 18 (4): 395–404. doi:10.1016/s0022-0736(85)80022-4. PMID 3906012.
- Childers R, Lupovich S, Sochanski M, Konarzewska H (2000). "Left bundle branch block and right axis deviation: a report of 36 cases". Journal of Electrocardiology. 33 Suppl (Suppl): 93–102. doi:10.1054/jclc.2000.20326. PMID 11265743.
- Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. (February 2012). "Truncations of titin causing dilated cardiomyopathy". The New England Journal of Medicine. 366 (7): 619–28. doi:10.1056/NEJMoa1110186. PMC 3660031. PMID 22335739.
- Pennell DJ, Sechtem UP, Higgins CB, Manning WJ, Pohost GM, Rademakers FE, et al. (November 2004). "Clinical indications for cardiovascular magnetic resonance (CMR): Consensus Panel report". European Heart Journal. 25 (21): 1940–65. doi:10.1016/j.ehj.2004.06.040. PMID 15522474.
- Langsjoen PH, Langsjoen PH, Folkers K (1990). "A six-year clinical study of therapy of cardiomyopathy with coenzyme Q10". International Journal of Tissue Reactions. 12 (3): 169–71. PMID 2276895.
- Folkers K, Langsjoen P, Langsjoen PH (January 1992). "Therapy with coenzyme Q10 of patients in heart failure who are eligible or ineligible for a transplant". Biochemical and Biophysical Research Communications. 182 (1): 247–53. doi:10.1016/S0006-291X(05)80137-8. PMID 1731784.
- Baggio E, Gandini R, Plancher AC, Passeri M, Carmosino G (1994). "Italian multicenter study on the safety and efficacy of coenzyme Q10 as adjunctive therapy in heart failure. CoQ10 Drug Surveillance Investigators". Molecular Aspects of Medicine. 15 Suppl (Suppl): s287-94. doi:10.1016/0098-2997(94)90040-X. PMID 7752841.
- Papadakis MA, McPhee SJ, Rabow MW (2016-09-01). Current medical diagnosis & treatment 2017 (Fifty-sixth ed.). New York. ISBN 978-1259585111. OCLC 957316517.
- Priori, Silvia G.; Blomström-Lundqvist, Carina; Mazzanti, Andrea; Blom, Nico; Borggrefe, Martin; Camm, John; Elliott, Perry Mark; Fitzsimons, Donna; Hatala, Robert; Hindricks, Gerhard; Kirchhof, Paulus (November 2015). "2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC)". Europace. 17 (11): 1601–1687. doi:10.1093/europace/euv319. ISSN 1532-2092. PMID 26318695.
- Kayvanpour E, Sammani A, Sedaghat-Hamedani F, Lehmann DH, Broezel A, Koelemenoglu J, Chmielewski P, Curjol A, Socie P, Miersch T, Haas J, Gi WT, Richard P, Płoski R, Truszkowska G, Baas AF, Foss-Nieradko B, Michalak E, Stępień-Wojno M, Zakrzewska-Koperska J, Śpiewak M, Zieliński T, Villard E, TeRiele ASJM, Katus HA, Frey N, Bilińska ZT, Charron P, Asselbergs FW, Meder B. A novel risk model for predicting potentially life-threatening arrhythmias in non-ischemic dilated cardiomyopathy (DCM-SVA risk). Int J Cardiol. 2021 Sep15;339:75-82. doi: 10.1016/j.ijcard.2021.07.002. Epub 2021 Jul 7. PMID 34245791.
- Coughlin SS, Labenberg JR, Tefft MC (March 1993). "Black-white differences in idiopathic dilated cardiomyopathy: the Washington DC dilated Cardiomyopathy Study". Epidemiology. 4 (2): 165–72. doi:10.1097/00001648-199303000-00013. PMID 8452906. S2CID 10961862.
- Reis Filho JR, Cardoso JN, Cardoso CM, Pereira-Barretto AC (June 2015). "Reverse Cardiac Remodeling: A Marker of Better Prognosis in Heart Failure". Arquivos Brasileiros de Cardiologia. 104 (6): 502–6. doi:10.5935/abc.20150025. PMC 4484683. PMID 26131706.
- Oyama MA, Chittur S (July 2005). "Genomic expression patterns of cardiac tissues from dogs with dilated cardiomyopathy". American Journal of Veterinary Research. 66 (7): 1140–55. doi:10.2460/ajvr.2005.66.1140. PMID 16111151.
- Quinton A (28 January 2019). "Dogs Fed Some Popular Diets Could Be at Risk of Heart Disease". UC Davis. Retrieved 2021-02-10.
- Freeman LM, Stern JA, Fries R, Adin DB, Rush JE (December 2018). "Diet-associated dilated cardiomyopathy in dogs: what do we know?". Journal of the American Veterinary Medical Association. 253 (11): 1390–1394. doi:10.2460/javma.253.11.1390. PMID 30451613.
- Kaplan JL, Stern JA, Fascetti AJ, Larsen JA, Skolnik H, Peddle GD, et al. (2018-12-13). "Taurine deficiency and dilated cardiomyopathy in golden retrievers fed commercial diets". PLOS ONE. 13 (12): e0209112. Bibcode:2018PLoSO..1309112K. doi:10.1371/journal.pone.0209112. PMC 6292607. PMID 30543707.
- Sanderson SL (November 2006). "Taurine and carnitine in canine cardiomyopathy". The Veterinary Clinics of North America. Small Animal Practice. 36 (6): 1325–43, vii–viii. doi:10.1016/j.cvsm.2006.08.010. PMID 17085238.
- Pion PD, Kittleson MD, Thomas WP, Skiles ML, Rogers QR (July 1992). "Clinical findings in cats with dilated cardiomyopathy and relationship of findings to taurine deficiency". Journal of the American Veterinary Medical Association. 201 (2): 267–74. PMID 1500323.
- Nigro V, Okazaki Y, Belsito A, Piluso G, Matsuda Y, Politano L, et al. (April 1997). "Identification of the Syrian hamster cardiomyopathy gene". Human Molecular Genetics. 6 (4): 601–7. doi:10.1093/hmg/6.4.601. PMID 9097966.
External links
- Dilated cardiomyopathy Archived 2013-05-25 at the Wayback Machine information for parents.