Drug metabolism
Drug metabolism is the metabolic breakdown of drugs by living organisms, usually through specialized enzymatic systems. More generally, xenobiotic metabolism (from the Greek xenos "stranger" and biotic "related to living beings") is the set of metabolic pathways that modify the chemical structure of xenobiotics, which are compounds foreign to an organism's normal biochemistry, such as any drug or poison. These pathways are a form of biotransformation present in all major groups of organisms and are considered to be of ancient origin. These reactions often act to detoxify poisonous compounds (although in some cases the intermediates in xenobiotic metabolism can themselves cause toxic effects). The study of drug metabolism is called pharmacokinetics.
The metabolism of pharmaceutical drugs is an important aspect of pharmacology and medicine. For example, the rate of metabolism determines the duration and intensity of a drug's pharmacologic action. Drug metabolism also affects multidrug resistance in infectious diseases and in chemotherapy for cancer, and the actions of some drugs as substrates or inhibitors of enzymes involved in xenobiotic metabolism are a common reason for hazardous drug interactions. These pathways are also important in environmental science, with the xenobiotic metabolism of microorganisms determining whether a pollutant will be broken down during bioremediation, or persist in the environment. The enzymes of xenobiotic metabolism, particularly the glutathione S-transferases are also important in agriculture, since they may produce resistance to pesticides and herbicides.
Drug metabolism is divided into three phases. In phase I, enzymes such as cytochrome P450 oxidases introduce reactive or polar groups into xenobiotics. These modified compounds are then conjugated to polar compounds in phase II reactions. These reactions are catalysed by transferase enzymes such as glutathione S-transferases. Finally, in phase III, the conjugated xenobiotics may be further processed, before being recognised by efflux transporters and pumped out of cells. Drug metabolism often converts lipophilic compounds into hydrophilic products that are more readily excreted.
Permeability barriers and detoxification
The exact compounds an organism is exposed to will be largely unpredictable, and may differ widely over time; these are major characteristics of xenobiotic toxic stress.[1] The major challenge faced by xenobiotic detoxification systems is that they must be able to remove the almost-limitless number of xenobiotic compounds from the complex mixture of chemicals involved in normal metabolism. The solution that has evolved to address this problem is an elegant combination of physical barriers and low-specificity enzymatic systems.
All organisms use cell membranes as hydrophobic permeability barriers to control access to their internal environment. Polar compounds cannot diffuse across these cell membranes, and the uptake of useful molecules is mediated through transport proteins that specifically select substrates from the extracellular mixture. This selective uptake means that most hydrophilic molecules cannot enter cells, since they are not recognised by any specific transporters.[2] In contrast, the diffusion of hydrophobic compounds across these barriers cannot be controlled, and organisms, therefore, cannot exclude lipid-soluble xenobiotics using membrane barriers.
However, the existence of a permeability barrier means that organisms were able to evolve detoxification systems that exploit the hydrophobicity common to membrane-permeable xenobiotics. These systems therefore solve the specificity problem by possessing such broad substrate specificities that they metabolise almost any non-polar compound.[1] Useful metabolites are excluded since they are polar, and in general contain one or more charged groups.
The detoxification of the reactive by-products of normal metabolism cannot be achieved by the systems outlined above, because these species are derived from normal cellular constituents and usually share their polar characteristics. However, since these compounds are few in number, specific enzymes can recognize and remove them. Examples of these specific detoxification systems are the glyoxalase system, which removes the reactive aldehyde methylglyoxal,[3] and the various antioxidant systems that eliminate reactive oxygen species.[4]
Phases of detoxification
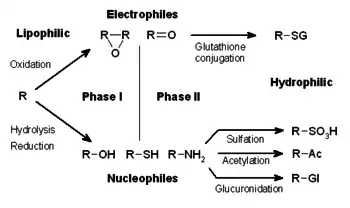
The metabolism of xenobiotics is often divided into three phases:- modification, conjugation, and excretion. These reactions act in concert to detoxify xenobiotics and remove them from cells.
Phase I – modification
In phase I, a variety of enzymes act to introduce reactive and polar groups into their substrates. One of the most common modifications is hydroxylation catalysed by the cytochrome P-450-dependent mixed-function oxidase system. These enzyme complexes act to incorporate an atom of oxygen into nonactivated hydrocarbons, which can result in either the introduction of hydroxyl groups or N-, O- and S-dealkylation of substrates.[5] The reaction mechanism of the P-450 oxidases proceeds through the reduction of cytochrome-bound oxygen and the generation of a highly-reactive oxyferryl species, according to the following scheme:[6]
- O2 + NADPH + H+ + RH → NADP+ + H2O + ROH
Phase I reactions (also termed nonsynthetic reactions) may occur by oxidation, reduction, hydrolysis, cyclization, decyclization, and addition of oxygen or removal of hydrogen, carried out by mixed function oxidases, often in the liver. These oxidative reactions typically involve a cytochrome P450 monooxygenase (often abbreviated CYP), NADPH and oxygen. The classes of pharmaceutical drugs that utilize this method for their metabolism include phenothiazines, paracetamol, and steroids. If the metabolites of phase I reactions are sufficiently polar, they may be readily excreted at this point. However, many phase I products are not eliminated rapidly and undergo a subsequent reaction in which an endogenous substrate combines with the newly incorporated functional group to form a highly polar conjugate.
A common Phase I oxidation involves conversion of a C-H bond to a C-OH. This reaction sometimes converts a pharmacologically inactive compound (a prodrug) to a pharmacologically active one. By the same token, Phase I can turn a nontoxic molecule into a poisonous one (toxification). Simple hydrolysis in the stomach is normally an innocuous reaction, however there are exceptions. For example, phase I metabolism converts acetonitrile to HOCH2CN, which rapidly dissociates into formaldehyde and hydrogen cyanide.[7]
Phase I metabolism of drug candidates can be simulated in the laboratory using non-enzyme catalysts.[8] This example of a biomimetic reaction tends to give products that often contains the Phase I metabolites. As an example, the major metabolite of the pharmaceutical trimebutine, desmethyltrimebutine (nor-trimebutine), can be efficiently produced by in vitro oxidation of the commercially available drug. Hydroxylation of an N-methyl group leads to expulsion of a molecule of formaldehyde, while oxidation of the O-methyl groups takes place to a lesser extent.
Oxidation
- Cytochrome P450 monooxygenase system
- Flavin-containing monooxygenase system
- Alcohol dehydrogenase and aldehyde dehydrogenase
- Monoamine oxidase
- Co-oxidation by peroxidases
Reduction
- NADPH-cytochrome P450 reductase
Cytochrome P450 reductase, also known as NADPH:ferrihemoprotein oxidoreductase, NADPH:hemoprotein oxidoreductase, NADPH:P450 oxidoreductase, P450 reductase, POR, CPR, CYPOR, is a membrane-bound enzyme required for electron transfer to cytochrome P450 in the microsome of the eukaryotic cell from a FAD- and FMN-containing enzyme NADPH:cytochrome P450 reductase The general scheme of electron flow in the POR/P450 system is: NADPH → FAD → FMN → P450 → O2
- Reduced (ferrous) cytochrome P450
During reduction reactions, a chemical can enter futile cycling, in which it gains a free-radical electron, then promptly loses it to oxygen (to form a superoxide anion).
Hydrolysis
- Esterases and amidase
- Epoxide hydrolase
Phase II – conjugation
In subsequent phase II reactions, these activated xenobiotic metabolites are conjugated with charged species such as glutathione (GSH), sulfate, glycine, or glucuronic acid. Sites on drugs where conjugation reactions occur include carboxy (-COOH), hydroxy (-OH), amino (NH2), and thiol (-SH) groups. Products of conjugation reactions have increased molecular weight and tend to be less active than their substrates, unlike Phase I reactions which often produce active metabolites. The addition of large anionic groups (such as GSH) detoxifies reactive electrophiles and produces more polar metabolites that cannot diffuse across membranes, and may, therefore, be actively transported.
These reactions are catalysed by a large group of broad-specificity transferases, which in combination can metabolise almost any hydrophobic compound that contains nucleophilic or electrophilic groups.[1] One of the most important classes of this group is that of the glutathione S-transferases (GSTs).
| Mechanism | Involved enzyme | Co-factor | Location | Sources |
|---|---|---|---|---|
| methylation | methyltransferase | S-adenosyl-L-methionine | liver, kidney, lung, CNS | [9] |
| sulphation | sulfotransferases | 3'-phosphoadenosine-5'-phosphosulfate | liver, kidney, intestine | [9] |
| acetylation |
|
acetyl coenzyme A | liver, lung, spleen, gastric mucosa, RBCs, lymphocytes | [9] |
| glucuronidation | UDP-glucuronosyltransferases | UDP-glucuronic acid | liver, kidney, intestine, lung, skin, prostate, brain | [9] |
| glutathione conjugation | glutathione S-transferases | glutathione | liver, kidney | [9] |
| glycine conjugation | Two step process:
|
glycine | liver, kidney | [10] |
Phase III – further modification and excretion
After phase II reactions, the xenobiotic conjugates may be further metabolized. A common example is the processing of glutathione conjugates to acetylcysteine (mercapturic acid) conjugates.[11] Here, the γ-glutamate and glycine residues in the glutathione molecule are removed by Gamma-glutamyl transpeptidase and dipeptidases. In the final step, the cysteine residue in the conjugate is acetylated.
Conjugates and their metabolites can be excreted from cells in phase III of their metabolism, with the anionic groups acting as affinity tags for a variety of membrane transporters of the multidrug resistance protein (MRP) family.[12] These proteins are members of the family of ATP-binding cassette transporters and can catalyse the ATP-dependent transport of a huge variety of hydrophobic anions,[13] and thus act to remove phase II products to the extracellular medium, where they may be further metabolized or excreted.[14]
Endogenous toxins
The detoxification of endogenous reactive metabolites such as peroxides and reactive aldehydes often cannot be achieved by the system described above. This is the result of these species' being derived from normal cellular constituents and usually sharing their polar characteristics. However, since these compounds are few in number, it is possible for enzymatic systems to utilize specific molecular recognition to recognize and remove them. The similarity of these molecules to useful metabolites therefore means that different detoxification enzymes are usually required for the metabolism of each group of endogenous toxins. Examples of these specific detoxification systems are the glyoxalase system, which acts to dispose of the reactive aldehyde methylglyoxal, and the various antioxidant systems that remove reactive oxygen species.
Sites
Quantitatively, the smooth endoplasmic reticulum of the liver cell is the principal organ of drug metabolism, although every biological tissue has some ability to metabolize drugs. Factors responsible for the liver's contribution to drug metabolism include that it is a large organ, that it is the first organ perfused by chemicals absorbed in the gut, and that there are very high concentrations of most drug-metabolizing enzyme systems relative to other organs. If a drug is taken into the GI tract, where it enters hepatic circulation through the portal vein, it becomes well-metabolized and is said to show the first pass effect.
Other sites of drug metabolism include epithelial cells of the gastrointestinal tract, lungs, kidneys, and the skin. These sites are usually responsible for localized toxicity reactions.
Factors that affect drug metabolism
The duration and intensity of pharmacological action of most lipophilic drugs are determined by the rate they are metabolized to inactive products. The Cytochrome P450 monooxygenase system is the most important pathway in this regard. In general, anything that increases the rate of metabolism (e.g., enzyme induction) of a pharmacologically active metabolite will decrease the duration and intensity of the drug action. The opposite is also true (e.g., enzyme inhibition). However, in cases where an enzyme is responsible for metabolizing a pro-drug into a drug, enzyme induction can speed up this conversion and increase drug levels, potentially causing toxicity.
Various physiological and pathological factors can also affect drug metabolism. Physiological factors that can influence drug metabolism include age, individual variation (e.g., pharmacogenetics), enterohepatic circulation, nutrition, intestinal flora, or sex differences.
In general, drugs are metabolized more slowly in fetal, neonatal and elderly humans and animals than in adults.
Genetic variation (polymorphism) accounts for some of the variability in the effect of drugs. With N-acetyltransferases (involved in Phase II reactions), individual variation creates a group of people who acetylate slowly (slow acetylators) and those who acetylate quickly, split roughly 50:50 in the population of Canada. This variation may have dramatic consequences, as the slow acetylators are more prone to dose-dependent toxicity.
Cytochrome P450 monooxygenase system enzymes can also vary across individuals, with deficiencies occurring in 1 – 30% of people, depending on their ethnic background.
Dose, frequency, route of administration, tissue distribution and protein binding of the drug affect its metabolism.
Pathological factors can also influence drug metabolism, including liver, kidney, or heart diseases.
In silico modelling and simulation methods allow drug metabolism to be predicted in virtual patient populations prior to performing clinical studies in human subjects.[15] This can be used to identify individuals most at risk from adverse reaction.
History
Studies on how people transform the substances that they ingest began in the mid-nineteenth century, with chemists discovering that organic chemicals such as benzaldehyde could be oxidized and conjugated to amino acids in the human body.[16] During the remainder of the nineteenth century, several other basic detoxification reactions were discovered, such as methylation, acetylation, and sulfonation.
In the early twentieth century, work moved on to the investigation of the enzymes and pathways that were responsible for the production of these metabolites. This field became defined as a separate area of study with the publication by Richard Williams of the book Detoxication mechanisms in 1947.[17] This modern biochemical research resulted in the identification of glutathione S-transferases in 1961,[18] followed by the discovery of cytochrome P450s in 1962,[19] and the realization of their central role in xenobiotic metabolism in 1963.[20][21]
See also
- Antioxidant
- Biodegradation
- Bioremediation
- Microbial biodegradation
References
- Jakoby WB, Ziegler DM (December 1990). "The enzymes of detoxication". J. Biol. Chem. 265 (34): 20715–8. doi:10.1016/S0021-9258(17)45272-0. PMID 2249981. Archived from the original on 2009-06-21. Retrieved 2012-12-29.
- Mizuno N, Niwa T, Yotsumoto Y, Sugiyama Y (September 2003). "Impact of drug transporter studies on drug discovery and development". Pharmacol. Rev. 55 (3): 425–61. doi:10.1124/pr.55.3.1. PMID 12869659. S2CID 724685.
- Thornalley PJ (July 1990). "The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life". Biochem. J. 269 (1): 1–11. doi:10.1042/bj2690001. PMC 1131522. PMID 2198020.
- Sies H (March 1997). "Oxidative stress: oxidants and antioxidants". Exp. Physiol. 82 (2): 291–5. doi:10.1113/expphysiol.1997.sp004024. PMID 9129943.
- Guengerich FP (June 2001). "Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity". Chem. Res. Toxicol. 14 (6): 611–50. doi:10.1021/tx0002583. PMID 11409933.
- Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG (March 2000). "The catalytic pathway of cytochrome p450cam at atomic resolution". Science. 287 (5458): 1615–22. Bibcode:2000Sci...287.1615S. doi:10.1126/science.287.5458.1615. PMID 10698731.
- "Acetonitrile (EHC 154, 1993)". www.inchem.org. Archived from the original on 2017-05-22. Retrieved 2017-05-03.
- Akagah B, Lormier AT, Fournet A, Figadère B (December 2008). "Oxidation of antiparasitic 2-substituted quinolines using metalloporphyrin catalysts: scale-up of a biomimetic reaction for metabolite production of drug candidates". Org. Biomol. Chem. 6 (24): 4494–7. doi:10.1039/b815963g. PMID 19039354.
- Liston HL, Markowitz JS, DeVane CL (October 2001). "Drug glucuronidation in clinical psychopharmacology". J Clin Psychopharmacol. 21 (5): 500–15. doi:10.1097/00004714-200110000-00008. PMID 11593076. S2CID 6068811.
- Badenhorst CP, van der Sluis R, Erasmus E, van Dijk AA (September 2013). "Glycine conjugation: importance in metabolism, the role of glycine N-acyltransferase, and factors that influence interindividual variation". Expert Opinion on Drug Metabolism & Toxicology. 9 (9): 1139–1153. doi:10.1517/17425255.2013.796929. PMID 23650932. S2CID 23738007.
Glycine conjugation of mitochondrial acyl-CoAs, catalyzed by glycine N-acyltransferase (GLYAT, E.C. 2.3.1.13), is an important metabolic pathway responsible for maintaining adequate levels of free coenzyme A (CoASH). However, because of the small number of pharmaceutical drugs that are conjugated to glycine, the pathway has not yet been characterized in detail. Here, we review the causes and possible consequences of interindividual variation in the glycine conjugation pathway. ...
Figure 1. Glycine conjugation of benzoic acid. The glycine conjugation pathway consists of two steps. First benzoate is ligated to CoASH to form the high-energy benzoyl-CoA thioester. This reaction is catalyzed by the HXM-A and HXM-B medium-chain acid:CoA ligases and requires energy in the form of ATP. ... The benzoyl-CoA is then conjugated to glycine by GLYAT to form hippuric acid, releasing CoASH. In addition to the factors listed in the boxes, the levels of ATP, CoASH, and glycine may influence the overall rate of the glycine conjugation pathway. - Boyland E, Chasseaud LF (1969). "The role of glutathione and glutathione S-transferases in mercapturic acid biosynthesis". Adv. Enzymol. Relat. Areas Mol. Biol. Advances in Enzymology – and Related Areas of Molecular Biology. 32: 173–219. doi:10.1002/9780470122778.ch5. ISBN 9780470122778. PMID 4892500.
- Homolya L, Váradi A, Sarkadi B (2003). "Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate". BioFactors. 17 (1–4): 103–14. doi:10.1002/biof.5520170111. PMID 12897433. S2CID 7744924.
- König J, Nies AT, Cui Y, Leier I, Keppler D (December 1999). "Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance". Biochim. Biophys. Acta. 1461 (2): 377–94. doi:10.1016/S0005-2736(99)00169-8. PMID 10581368.
- Commandeur JN, Stijntjes GJ, Vermeulen NP (June 1995). "Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics". Pharmacol. Rev. 47 (2): 271–330. PMID 7568330.
- Rostami-Hodjegan A, Tucker GT (February 2007). "Simulation and prediction of in vivo drug metabolism in human populations from in vitro data". Nat Rev Drug Discov. 6 (2): 140–8. doi:10.1038/nrd2173. PMID 17268485. S2CID 205476485.
- Murphy PJ (June 2001). "Xenobiotic metabolism: a look from the past to the future". Drug Metab. Dispos. 29 (6): 779–80. PMID 11353742. Archived from the original on 2009-06-21. Retrieved 2012-12-29.
- Neuberger A, Smith RL (1983). "Richard Tecwyn Williams: the man, his work, his impact". Drug Metab. Rev. 14 (3): 559–607. doi:10.3109/03602538308991399. PMID 6347595.
- Booth J, Boyland E, Sims P (June 1961). "An enzyme from rat liver catalysing conjugations with glutathione". Biochem. J. 79 (3): 516–24. doi:10.1042/bj0790516. PMC 1205680. PMID 16748905.
- Omura T, Sato R (April 1962). "A new cytochrome in liver microsomes". J. Biol. Chem. 237 (4): 1375–6. doi:10.1016/S0021-9258(18)60338-2. PMID 14482007. Archived from the original on 2009-06-21. Retrieved 2012-12-29.
- Estabrook RW (December 2003). "A passion for P450s (remembrances of the early history of research on cytochrome P450)". Drug Metab. Dispos. 31 (12): 1461–73. doi:10.1124/dmd.31.12.1461. PMID 14625342.
- Estabrook RW, Cooper DY, Rosenthal O (1963). "The light reversible carbon monoxide inhibition of steroid C-21 hydroxylase system in adrenal cortex". Biochem Z. 338: 741–55. PMID 14087340.
Further reading
- Parvez H, Reiss C (2001). Molecular Responses to Xenobiotics. Elsevier. ISBN 0-345-42277-5.
- Ioannides C (2001). Enzyme Systems That Metabolise Drugs and Other Xenobiotics. John Wiley and Sons. ISBN 0-471-89466-4.
- Richardson M (1996). Environmental Xenobiotics. Taylor & Francis Ltd. ISBN 0-7484-0399-X.
- Ioannides C (1996). Cytochromes P450: Metabolic and Toxicological Aspects. CRC Press Inc. ISBN 0-8493-9224-1.
- Awasthi YC (2006). Toxicology of Glutathionine S-transferses. CRC Press Inc. ISBN 0-8493-2983-3.
External links
- Databases
- Drug metabolism
- Microbial biodegradation
- History
- History of Xenobiotic Metabolism at the Wayback Machine (archived July 13, 2007)