Glass transition
The glass–liquid transition, or glass transition, is the gradual and reversible transition in amorphous materials (or in amorphous regions within semicrystalline materials) from a hard and relatively brittle "glassy" state into a viscous or rubbery state as the temperature is increased.[1][2] An amorphous solid that exhibits a glass transition is called a glass. The reverse transition, achieved by supercooling a viscous liquid into the glass state, is called vitrification.
The glass-transition temperature Tg of a material characterizes the range of temperatures over which this glass transition occurs. It is always lower than the melting temperature, Tm, of the crystalline state of the material, if one exists.
Hard plastics like polystyrene and poly(methyl methacrylate) are used well below their glass transition temperatures, i.e., when they are in their glassy state. Their Tg values are both at around 100 °C (212 °F). Rubber elastomers like polyisoprene and polyisobutylene are used above their Tg, that is, in the rubbery state, where they are soft and flexible; crosslinking prevents free flow of their molecules, thus endowing rubber with a set shape at room temperature (as opposed to a viscous liquid).[3]
Despite the change in the physical properties of a material through its glass transition, the transition is not considered a phase transition; rather it is a phenomenon extending over a range of temperature and defined by one of several conventions.[2][4][5] Such conventions include a constant cooling rate (20 kelvins per minute (36 °F/min))[1] and a viscosity threshold of 1012 Pa·s, among others. Upon cooling or heating through this glass-transition range, the material also exhibits a smooth step in the thermal-expansion coefficient and in the specific heat, with the location of these effects again being dependent on the history of the material.[6] The question of whether some phase transition underlies the glass transition is a matter of ongoing research.[4][5][7]
Glass transition (in polymer science): process in which a polymer melt changes on cooling to a polymer glass or a polymer glass changes on heating to a polymer melt.[8]
- Phenomena occurring at the glass transition of polymers are still subject to ongoing scientific investigation and debate. The glass transition presents features of a second-order
transition since thermal studies often indicate that the molar Gibbs energies, molar enthalpies, and the molar volumes of the two phases, i.e., the melt and the glass, are equal, while the heat capacity and the expansivity are discontinuous. However, the glass transition is generally not regarded as a thermodynamic transition in view of the inherent difficulty in reaching equilibrium in a polymer glass or in a polymer melt at temperatures close to the glass-transition temperature.
- In the case of polymers, conformational changes of segments, typically consisting of 10–20 main-chain atoms, become infinitely slow below the glass transition temperature.
- In a partially crystalline polymer the glass transition occurs only in the amorphous parts of the material.
- The definition is different from that in ref.[9]
- The commonly used term “glass-rubber transition” for glass transition is not recommended.[8]
Introduction
The glass transition of a liquid to a solid-like state may occur with either cooling or compression.[10] The transition comprises a smooth increase in the viscosity of a material by as much as 17 orders of magnitude within a temperature range of 500 K without any pronounced change in material structure.[2][11] The consequence of this dramatic increase is a glass exhibiting solid-like mechanical properties on the timescale of practical observation. This transition is in contrast to the freezing or crystallization transition, which is a first-order phase transition in the Ehrenfest classification and involves discontinuities in thermodynamic and dynamic properties such as volume, energy, and viscosity. In many materials that normally undergo a freezing transition, rapid cooling will avoid this phase transition and instead result in a glass transition at some lower temperature. Other materials, such as many polymers, lack a well defined crystalline state and easily form glasses, even upon very slow cooling or compression. The tendency for a material to form a glass while quenched is called glass forming ability. This ability depends on the composition of the material and can be predicted by the rigidity theory.[12]
Below the transition temperature range, the glassy structure does not relax in accordance with the cooling rate used. The expansion coefficient for the glassy state is roughly equivalent to that of the crystalline solid. If slower cooling rates are used, the increased time for structural relaxation (or intermolecular rearrangement) to occur may result in a higher density glass product. Similarly, by annealing (and thus allowing for slow structural relaxation) the glass structure in time approaches an equilibrium density corresponding to the supercooled liquid at this same temperature. Tg is located at the intersection between the cooling curve (volume versus temperature) for the glassy state and the supercooled liquid.[2][13][14][15][16][17]
The configuration of the glass in this temperature range changes slowly with time towards the equilibrium structure.[18] The principle of the minimization of the Gibbs free energy provides the thermodynamic driving force necessary for the eventual change. At somewhat higher temperatures than Tg, the structure corresponding to equilibrium at any temperature is achieved quite rapidly. In contrast, at considerably lower temperatures, the configuration of the glass remains sensibly stable over increasingly extended periods of time.
Thus, the liquid-glass transition is not a transition between states of thermodynamic equilibrium. It is widely believed that the true equilibrium state is always crystalline. Glass is believed to exist in a kinetically locked state, and its entropy, density, and so on, depend on the thermal history. Therefore, the glass transition is primarily a dynamic phenomenon. Time and temperature are interchangeable quantities (to some extent) when dealing with glasses, a fact often expressed in the time–temperature superposition principle. On cooling a liquid, internal degrees of freedom successively fall out of equilibrium. However, there is a longstanding debate whether there is an underlying second-order phase transition in the hypothetical limit of infinitely long relaxation times.[6][19][20][21]
In a more recent model of glass transition, the glass transition temperature corresponds to the temperature at which the largest openings between the vibrating elements in the liquid matrix become smaller than the smallest cross-sections of the elements or parts of them when the temperature is decreasing. As a result of the fluctuating input of thermal energy into the liquid matrix, the harmonics of the oscillations are constantly disturbed and temporary cavities ("free volume") are created between the elements, the number and size of which depend on the temperature. The glass transition temperature Tg0 defined in this way is a fixed material constant of the disordered (non-crystalline) state that is dependent only on the pressure. As a result of the increasing inertia of the molecular matrix when approaching Tg0, the setting of the thermal equilibrium is successively delayed, so that the usual measuring methods for determining the glass transition temperature in principle deliver Tg values that are too high. In principle, the slower the temperature change rate is set during the measurement, the closer the measured Tg value Tg0 approaches.[22] Techniques such as dynamic mechanical analysis can be used to measure the glass transition temperature.[23]
Transition temperature Tg

Refer to the figure on the bottom right plotting the heat capacity as a function of temperature. In this context, Tg is the temperature corresponding to point A on the curve.[24]
Different operational definitions of the glass transition temperature Tg are in use, and several of them are endorsed as accepted scientific standards. Nevertheless, all definitions are arbitrary, and all yield different numeric results: at best, values of Tg for a given substance agree within a few kelvins. One definition refers to the viscosity, fixing Tg at a value of 1013 poise (or 1012 Pa·s). As evidenced experimentally, this value is close to the annealing point of many glasses.[25]
In contrast to viscosity, the thermal expansion, heat capacity, shear modulus, and many other properties of inorganic glasses show a relatively sudden change at the glass transition temperature. Any such step or kink can be used to define Tg. To make this definition reproducible, the cooling or heating rate must be specified.
The most frequently used definition of Tg uses the energy release on heating in differential scanning calorimetry (DSC, see figure). Typically, the sample is first cooled with 10 K/min and then heated with that same speed.
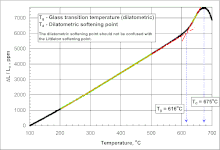
Yet another definition of Tg uses the kink in dilatometry (a.k.a. thermal expansion): refer to the figure on the top right. Here, heating rates of 3–5 K/min (5.4–9.0 °F/min) are common. The linear sections below and above Tg are colored green. Tg is the temperature at the intersection of the red regression lines.[24]
Summarized below are Tg values characteristic of certain classes of materials.
Polymers
| Material | Tg (°C) | Tg (°F) | Commercial name |
|---|---|---|---|
| Tire rubber | −70 | −94[26] | |
| Polyvinylidene fluoride (PVDF) | −35 | −31[27] | |
| Polypropylene (PP atactic) | −20 | −4[28] | |
| Polyvinyl fluoride (PVF) | −20 | −4[27] | |
| Polypropylene (PP isotactic) | 0 | 32[28] | |
| Poly-3-hydroxybutyrate (PHB) | 15 | 59[28] | |
| Poly(vinyl acetate) (PVAc) | 30 | 86[28] | |
| Polychlorotrifluoroethylene (PCTFE) | 45 | 113[27] | |
| Polyamide (PA) | 47–60 | 117–140 | Nylon-6,x |
| Polylactic acid (PLA) | 60–65 | 140–149 | |
| Polyethylene terephthalate (PET) | 70 | 158[28] | |
| Poly(vinyl chloride) (PVC) | 80 | 176[28] | |
| Poly(vinyl alcohol) (PVA) | 85 | 185[28] | |
| Polystyrene (PS) | 95 | 203[28] | |
| Poly(methyl methacrylate) (PMMA atactic) | 105 | 221[28] | Plexiglas, Perspex |
| Acrylonitrile butadiene styrene (ABS) | 105 | 221[29] | |
| Polytetrafluoroethylene (PTFE) | 115 | 239[30] | Teflon |
| Poly(carbonate) (PC) | 145 | 293[28] | Lexan |
| Polysulfone | 185 | 365 | |
| Polynorbornene | 215 | 419[28] |
Dry nylon-6 has a glass transition temperature of 47 °C (117 °F).[31] Nylon-6,6 in the dry state has a glass transition temperature of about 70 °C (158 °F).[32][33] Whereas polyethene has a glass transition range of −130 – −80 °C (−202 – −112 °F)[34] The above are only mean values, as the glass transition temperature depends on the cooling rate and molecular weight distribution and could be influenced by additives. For a semi-crystalline material, such as polyethene that is 60–80% crystalline at room temperature, the quoted glass transition refers to what happens to the amorphous part of the material upon cooling.
Silicates and other covalent network glasses
| Material | Tg (°C) | Tg (°F) |
|---|---|---|
| Chalcogenide GeSbTe | 150 | 302[35] |
| Chalcogenide AsGeSeTe | 245 | 473 |
| ZBLAN fluoride glass | 235 | 455 |
| Tellurium dioxide | 280 | 536 |
| Fluoroaluminate | 400 | 752 |
| Soda-lime glass | 520–600 | 968–1,112 |
| Fused quartz (approximate) | 1,200 | 2,200[36] |
Kauzmann's paradox
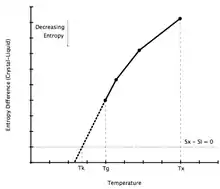
As a liquid is supercooled, the difference in entropy between the liquid and solid phase decreases. By extrapolating the heat capacity of the supercooled liquid below its glass transition temperature, it is possible to calculate the temperature at which the difference in entropies becomes zero. This temperature has been named the Kauzmann temperature.[2]
If a liquid could be supercooled below its Kauzmann temperature, and it did indeed display a lower entropy than the crystal phase, the consequences would be paradoxical. This Kauzmann paradox has been the subject of much debate and many publications since it was first put forward by Walter Kauzmann in 1948.[37][38]
One resolution of the Kauzmann paradox is to say that there must be a phase transition before the entropy of the liquid decreases. In this scenario, the transition temperature is known as the calorimetric ideal glass transition temperature T0c. In this view, the glass transition is not merely a kinetic effect, i.e. merely the result of fast cooling of a melt, but there is an underlying thermodynamic basis for glass formation. The glass transition temperature:

The Gibbs–DiMarzio model from 1958[39] specifically predicts that a supercooled liquid's configurational entropy disappears in the limit , where the liquid's existence regime ends, its microstructure becomes identical to the crystal's, and their property curves intersect in a true second-order phase transition. This has never been experimentally verified due to the difficulty of realizing a slow enough cooling rate while avoiding accidental crystallization. The Adam–Gibbs model from 1965[40] suggested a resolution of the Kauzmann paradox according to which the relaxation time diverges at the Kauzmann temperature, implying that one can never equilibrate the metastable supercooled liquid here. A critical discussion of the Kauzmann paradox and the Adam–Gibbs model was given in 2009.[41] Data on several supercooled organic liquids do not confirm the Adam–Gibbs prediction of a diverging relaxation time at any finite temperature, e.g. the Kauzmann temperature.[42]

Alternative resolutions
There are at least three other possible resolutions to the Kauzmann paradox. It could be that the heat capacity of the supercooled liquid near the Kauzmann temperature smoothly decreases to a smaller value. It could also be that a first order phase transition to another liquid state occurs before the Kauzmann temperature with the heat capacity of this new state being less than that obtained by extrapolation from higher temperature. Finally, Kauzmann himself resolved the entropy paradox by postulating that all supercooled liquids must crystallize before the Kauzmann temperature is reached.
In specific materials
Silica, SiO2
Silica (the chemical compound SiO2) has a number of distinct crystalline forms in addition to the quartz structure. Nearly all of the crystalline forms involve tetrahedral SiO4 units linked together by shared vertices in different arrangements (stishovite, composed of linked SiO6 octahedra, is the main exception). Si-O bond lengths vary between the different crystal forms. For example, in α-quartz the bond length is 161 picometres (6.3×10−9 in), whereas in α-tridymite it ranges from 154–171 pm (6.1×10−9–6.7×10−9 in). The Si-O-Si bond angle also varies from 140° in α-tridymite to 144° in α-quartz to 180° in β-tridymite. Any deviations from these standard parameters constitute microstructural differences or variations that represent an approach to an amorphous, vitreous or glassy solid. The transition temperature Tg in silicates is related to the energy required to break and re-form covalent bonds in an amorphous (or random network) lattice of covalent bonds. The Tg is clearly influenced by the chemistry of the glass. For example, addition of elements such as B, Na, K or Ca to a silica glass, which have a valency less than 4, helps in breaking up the network structure, thus reducing the Tg. Alternatively, P, which has a valency of 5, helps to reinforce an ordered lattice, and thus increases the Tg.[43] Tg is directly proportional to bond strength, e.g. it depends on quasi-equilibrium thermodynamic parameters of the bonds e.g. on the enthalpy Hd and entropy Sd of configurons – broken bonds: Tg = Hd / [Sd + R ln[(1 − fc)/ fc] where R is the gas constant and fc is the percolation threshold. For strong melts such as SiO2 the percolation threshold in the above equation is the universal Scher–Zallen critical density in the 3-D space e.g. fc = 0.15, however for fragile materials the percolation thresholds are material-dependent and fc ≪ 1.[44] The enthalpy Hd and the entropy Sd of configurons – broken bonds can be found from available experimental data on viscosity.[45]
Polymers
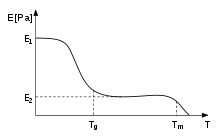
In polymers the glass transition temperature, Tg, is often expressed as the temperature at which the Gibbs free energy is such that the activation energy for the cooperative movement of 50 or so elements of the polymer is exceeded . This allows molecular chains to slide past each other when a force is applied. From this definition, we can see that the introduction of relatively stiff chemical groups (such as benzene rings) will interfere with the flowing process and hence increase Tg.[46] The stiffness of thermoplastics decreases due to this effect (see figure.) When the glass temperature has been reached, the stiffness stays the same for a while, i.e., at or near E2, until the temperature exceeds Tm, and the material melts. This region is called the rubber plateau.

Coming from the low-temperature side, the shear modulus drops by many orders of magnitude at the glass transition temperature Tg. A molecular-level mathematical relation for the temperature-dependent shear modulus of the polymer glass on approaching Tg from below has been developed by Alessio Zaccone and Eugene Terentjev.[47] Even though the shear modulus does not really drop to zero (it drops down to the much lower value of the rubber plateau), upon setting the shear modulus to zero in the Zaccone–Terentjev formula, an expression for Tg is obtained which recovers the Flory–Fox equation, and also shows that Tg is inversely proportional to the thermal expansion coefficient in the glass state. This procedure provides yet another operational protocol to define the Tg of polymer glasses by identifying it with the temperature at which the shear modulus drops by many orders of magnitude down to the rubbery plateau.
In ironing, a fabric is heated through this transition so that the polymer chains become mobile. The weight of the iron then imposes a preferred orientation. Tg can be significantly decreased by addition of plasticizers into the polymer matrix. Smaller molecules of plasticizer embed themselves between the polymer chains, increasing the spacing and free volume, and allowing them to move past one another even at lower temperatures. Addition of plasticizer can effectively take control over polymer chain dynamics and dominate the amounts of the associated free volume so that the increased mobility of polymer ends is not apparent.[48] The addition of nonreactive side groups to a polymer can also make the chains stand off from one another, reducing Tg. If a plastic with some desirable properties has a Tg that is too high, it can sometimes be combined with another in a copolymer or composite material with a Tg below the temperature of intended use. Note that some plastics are used at high temperatures, e.g., in automobile engines, and others at low temperatures.[28]
In viscoelastic materials, the presence of liquid-like behavior depends on the properties of and so varies with rate of applied load, i.e., how quickly a force is applied. The silicone toy Silly Putty behaves quite differently depending on the time rate of applying a force: pull slowly and it flows, acting as a heavily viscous liquid; hit it with a hammer and it shatters, acting as a glass.
On cooling, rubber undergoes a liquid-glass transition, which has also been called a rubber-glass transition.
Mechanics of vitrification
Molecular motion in condensed matter can be represented by a Fourier series whose physical interpretation consists of a superposition of longitudinal and transverse waves of atomic displacement with varying directions and wavelengths. In monatomic systems, these waves are called density fluctuations. (In polyatomic systems, they may also include compositional fluctuations.)[49]
Thus, thermal motion in liquids can be decomposed into elementary longitudinal vibrations (or acoustic phonons) while transverse vibrations (or shear waves) were originally described only in elastic solids exhibiting the highly ordered crystalline state of matter. In other words, simple liquids cannot support an applied force in the form of a shearing stress, and will yield mechanically via macroscopic plastic deformation (or viscous flow). Furthermore, the fact that a solid deforms locally while retaining its rigidity – while a liquid yields to macroscopic viscous flow in response to the application of an applied shearing force – is accepted by many as the mechanical distinction between the two.[50][51]
The inadequacies of this conclusion, however, were pointed out by Frenkel in his revision of the kinetic theory of solids and the theory of elasticity in liquids. This revision follows directly from the continuous characteristic of the viscoelastic crossover from the liquid state into the solid one when the transition is not accompanied by crystallization—ergo the supercooled viscous liquid. Thus we see the intimate correlation between transverse acoustic phonons (or shear waves) and the onset of rigidity upon vitrification, as described by Bartenev in his mechanical description of the vitrification process.[52][53] This concept leads to defining the glass transition in terms of the vanishing or significant lowering of the low-frequency shear modulus, as shown quantitatively in the work of Zaccone and Terentjev[47] on the example of polymer glass. In fact, the shoving model stipulates that the activation energy of the relaxation time is proportional to the high-frequency plateau shear modulus,[2][54] a quantity that increases upon cooling thus explaining the ubiquitous non-Arrhenius temperature dependence of the relaxation time in glass-forming liquids.
The velocities of longitudinal acoustic phonons in condensed matter are directly responsible for the thermal conductivity that levels out temperature differentials between compressed and expanded volume elements. Kittel proposed that the behavior of glasses is interpreted in terms of an approximately constant "mean free path" for lattice phonons, and that the value of the mean free path is of the order of magnitude of the scale of disorder in the molecular structure of a liquid or solid. The thermal phonon mean free paths or relaxation lengths of a number of glass formers have been plotted versus the glass transition temperature, indicating a linear relationship between the two. This has suggested a new criterion for glass formation based on the value of the phonon mean free path.[55]
It has often been suggested that heat transport in dielectric solids occurs through elastic vibrations of the lattice, and that this transport is limited by elastic scattering of acoustic phonons by lattice defects (e.g. randomly spaced vacancies).[56] These predictions were confirmed by experiments on commercial glasses and glass ceramics, where mean free paths were apparently limited by "internal boundary scattering" to length scales of 10–100 micrometres (0.00039–0.00394 in).[57][58] The relationship between these transverse waves and the mechanism of vitrification has been described by several authors who proposed that the onset of correlations between such phonons results in an orientational ordering or "freezing" of local shear stresses in glass-forming liquids, thus yielding the glass transition.[59]
Electronic structure
The influence of thermal phonons and their interaction with electronic structure is a topic that was appropriately introduced in a discussion of the resistance of liquid metals. Lindemann's theory of melting is referenced,[60] and it is suggested that the drop in conductivity in going from the crystalline to the liquid state is due to the increased scattering of conduction electrons as a result of the increased amplitude of atomic vibration. Such theories of localization have been applied to transport in metallic glasses, where the mean free path of the electrons is very small (on the order of the interatomic spacing).[61][62]
The formation of a non-crystalline form of a gold-silicon alloy by the method of splat quenching from the melt led to further considerations of the influence of electronic structure on glass forming ability, based on the properties of the metallic bond.[63][64][65][66][67]
Other work indicates that the mobility of localized electrons is enhanced by the presence of dynamic phonon modes. One claim against such a model is that if chemical bonds are important, the nearly free electron models should not be applicable. However, if the model includes the buildup of a charge distribution between all pairs of atoms just like a chemical bond (e.g., silicon, when a band is just filled with electrons) then it should apply to solids.[68]
Thus, if the electrical conductivity is low, the mean free path of the electrons is very short. The electrons will only be sensitive to the short-range order in the glass since they do not get a chance to scatter from atoms spaced at large distances. Since the short-range order is similar in glasses and crystals, the electronic energies should be similar in these two states. For alloys with lower resistivity and longer electronic mean free paths, the electrons could begin to sense that there is disorder in the glass, and this would raise their energies and destabilize the glass with respect to crystallization. Thus, the glass formation tendencies of certain alloys may therefore be due in part to the fact that the electron mean free paths are very short, so that only the short-range order is ever important for the energy of the electrons.
It has also been argued that glass formation in metallic systems is related to the "softness" of the interaction potential between unlike atoms. Some authors, emphasizing the strong similarities between the local structure of the glass and the corresponding crystal, suggest that chemical bonding helps to stabilize the amorphous structure.[69][70]
Other authors have suggested that the electronic structure yields its influence on glass formation through the directional properties of bonds. Non-crystallinity is thus favored in elements with a large number of polymorphic forms and a high degree of bonding anisotropy. Crystallization becomes more unlikely as bonding anisotropy is increased from isotropic metallic to anisotropic metallic to covalent bonding, thus suggesting a relationship between the group number in the periodic table and the glass forming ability in elemental solids.[71]
See also
- Gardner transition
References
- ISO 11357-2: Plastics – Differential scanning calorimetry – Part 2: Determination of glass transition temperature (1999).
- Dyre, Jeppe C. (2006). "Colloquium : The glass transition and elastic models of glass-forming liquids". Reviews of Modern Physics. 78 (3): 953–972. Bibcode:2006RvMP...78..953D. doi:10.1103/RevModPhys.78.953. ISSN 0034-6861.
- "The Glass Transition". Polymer Science Learning Center. Archived from the original on 2019-01-15. Retrieved 2009-10-15.
- Debenedetti, P. G.; Stillinger (2001). "Supercooled liquids and the glass transition". Nature. 410 (6825): 259–267. Bibcode:2001Natur.410..259D. doi:10.1038/35065704. PMID 11258381. S2CID 4404576.
- Angell, C. A.; Ngai, K. L.; McKenna, G. B.; McMillan, P. F.; Martin, S. W. (2000). "Relaxation in glassforming liquids and amorphous solids". Appl. Phys. Rev. 88 (6): 3113–3157. Bibcode:2000JAP....88.3113A. doi:10.1063/1.1286035. Archived from the original on 2020-03-07. Retrieved 2018-09-06.
- Zarzycki, J. (1991). Glasses and the Vitreous State. Cambridge University Press. ISBN 978-0521355827. Archived from the original on 2020-08-02. Retrieved 2016-09-23.
- Ojovan, M. I. (2004). "Glass formation in amorphous SiO2 as a percolation phase transition in a system of network defects". Journal of Experimental and Theoretical Physics Letters. 79 (12): 632–634. Bibcode:2004JETPL..79..632O. doi:10.1134/1.1790021. S2CID 124299526.
- Meille Stefano, V.; Allegra, G.; Geil Phillip, H.; He, J.; Hess, M.; Jin, J.-I.; Kratochvíl, P.; Mormann, W.; Stepto, R. (2011). "Definitions of terms relating to crystalline polymers (IUPAC Recommendations 2011)" (PDF). Pure and Applied Chemistry. 83 (10): 1831. doi:10.1351/PAC-REC-10-11-13. S2CID 98823962. Archived (PDF) from the original on 2018-06-25. Retrieved 2018-06-25.
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "glass transition". doi:10.1351/goldbook.G02640
- Hansen, J.-P.; McDonald, I. R. (2007). Theory of Simple Liquids. Elsevier. pp. 250–254. ISBN 978-0123705358. Archived from the original on 2013-09-21. Retrieved 2016-09-23.
- Adam, J-L; Zhang, X. (14 February 2014). Chalcogenide Glasses: Preparation, Properties and Applications. Elsevier Science. p. 94. ISBN 978-0-85709-356-1. Archived from the original on 17 July 2017. Retrieved 2 May 2017.
- Phillips, J.C. (1979). "Topology of covalent non-crystalline solids I: Short-range order in chalcogenide alloys". Journal of Non-Crystalline Solids. 34 (2): 153. Bibcode:1979JNCS...34..153P. doi:10.1016/0022-3093(79)90033-4.
- Moynihan, C. et al. (1976) in The Glass Transition and the Nature of the Glassy State, M. Goldstein and R. Simha (Eds.), Ann. N.Y. Acad. Sci., Vol. 279. ISBN 0890720533.
- Angell, C. A. (1988). "Perspective on the glass transition". Journal of Physics and Chemistry of Solids. 49 (8): 863–871. Bibcode:1988JPCS...49..863A. doi:10.1016/0022-3697(88)90002-9.
- Ediger, M. D.; Angell, C. A.; Nagel, Sidney R. (1996). "Supercooled Liquids and Glasses". The Journal of Physical Chemistry. 100 (31): 13200. doi:10.1021/jp953538d.
- Angell, C. A. (1995). "Formation of Glasses from Liquids and Biopolymers". Science. 267 (5206): 1924–35. Bibcode:1995Sci...267.1924A. doi:10.1126/science.267.5206.1924. PMID 17770101. S2CID 927260.
- Stillinger, F. H. (1995). "A Topographic View of Supercooled Liquids and Glass Formation". Science. 267 (5206): 1935–9. Bibcode:1995Sci...267.1935S. doi:10.1126/science.267.5206.1935. PMID 17770102. S2CID 30407650.
- Riechers, Birte; Roed, Lisa A.; Mehri, Saeed; Ingebrigtsen, Trond S.; Hecksher, Tina; Dyre, Jeppe C.; Niss, Kristine (2022-03-18). "Predicting nonlinear physical aging of glasses from equilibrium relaxation via the material time". Science Advances. 8 (11): eabl9809. arXiv:2109.11832. Bibcode:2022SciA....8L9809R. doi:10.1126/sciadv.abl9809. ISSN 2375-2548. PMC 8926348. PMID 35294250.
- Nemilov SV (1994). Thermodynamic and Kinetic Aspects of the Vitreous State. CRC Press. ISBN 978-0849337826.
- Gibbs, J. H. (1960). MacKenzie, J. D. (ed.). Modern Aspects of the Vitreous State. Butterworth. OCLC 1690554.
- Ojovan, Michael I; Lee, William (Bill) E (2010). "Connectivity and glass transition in disordered oxide systems". Journal of Non-Crystalline Solids. 356 (44–49): 2534. Bibcode:2010JNCS..356.2534O. doi:10.1016/j.jnoncrysol.2010.05.012.
- Sturm, Karl Günter (2017). "Microscopic-Phenomenological Model of Glass Transition I. Foundations of the model (Revised and enhanced version) (Former title: Microscopic Model of Glass Transformation and Molecular Translations in Liquids I. Foundations of the Model-October 2015)". doi:10.13140/RG.2.2.19831.73121.
{{cite journal}}: Cite journal requires|journal=(help) - "What is Dynamic Mechanical Testing (DMA)?". 2018. Archived from the original on 2020-11-17. Retrieved 2020-12-09.
- Tg measurement of glasses Archived 2009-04-17 at the Wayback Machine. Glassproperties.com. Retrieved on 2012-06-29.
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "glass-transition temperature". doi:10.1351/goldbook.G02641
- Galimberti, Maurizio; Caprio, Michela; Fino, Luigi (2001-12-21). "Tyre comprising a cycloolefin polymer, tread band and elasomeric composition used therein" (published 2003-03-07). Archived from the original on 2013-09-13. Retrieved 2013-09-10.
country-code =EU, patent-number =WO03053721
{{cite journal}}: Cite journal requires|journal=(help) - Ibeh, Christopher C. (2011). THERMOPLASTIC MATERIALS Properties, Manufacturing Methods, and Applications. CRC Press. pp. 491–497. ISBN 978-1-4200-9383-4.
- Wilkes, C. E. (2005). PVC Handbook. Hanser Verlag. ISBN 978-1-56990-379-7. Archived from the original on 2022-04-03. Retrieved 2016-09-23.
- ABS. nrri.umn.edu
- Nicholson, John W. (2011). The Chemistry of Polymers (4, Revised ed.). Royal Society of Chemistry. p. 50. ISBN 9781849733915. Archived from the original on 10 April 2022. Retrieved 10 September 2013.
- nylon-6 information and properties Archived 2012-01-10 at the Wayback Machine. Polymerprocessing.com (2001-04-15). Retrieved on 2012-06-29.
- Jones, A (2014). "Supplementary Materials for Artificial Muscles from Fishing Line and Sewing Thread". Science. 343 (6173): 868–72. Bibcode:2014Sci...343..868H. doi:10.1126/science.1246906. PMID 24558156. S2CID 16577662.
- Measurement of Moisture Effects on the Mechanical Properties of 66 Nylon. TA Instruments Thermal Analysis Application Brief TA-133
- PCL | Applications and End Uses | Polythene Archived 2013-06-05 at the Wayback Machine. Polyesterconverters.com. Retrieved on 2012-06-29.
- EPCOS 2007: Glass Transition and Crystallization in Phase Change Materials Archived 2011-07-26 at the Wayback Machine . Retrieved on 2012-06-29.
- Bucaro, J. A. (1974). "High-temperature Brillouin scattering in fused quartz". Journal of Applied Physics. 45 (12): 5324–5329. Bibcode:1974JAP....45.5324B. doi:10.1063/1.1663238.
- Kauzmann, Walter (1948). "The Nature of the Glassy State and the Behavior of Liquids at Low Temperatures". Chemical Reviews. 43 (2): 219–256. doi:10.1021/cr60135a002.
- Wolchover, Natalie (11 March 2020). "Ideal Glass Would Explain Why Glass Exists at All". Quanta Magazine. Archived from the original on 7 April 2020. Retrieved 3 April 2020.
- Gibbs, Julian H.; DiMarzio, Edmund A. (1958). "Nature of the Glass Transition and the Glassy State". The Journal of Chemical Physics. 28 (3): 373–383. Bibcode:1958JChPh..28..373G. doi:10.1063/1.1744141. ISSN 0021-9606. S2CID 97800386.
- Adam, Gerold; Gibbs, Julian H. (1965). "On the Temperature Dependence of Cooperative Relaxation Properties in Glass‐Forming Liquids". The Journal of Chemical Physics. 43 (1): 139–146. Bibcode:1965JChPh..43..139A. doi:10.1063/1.1696442. ISSN 0021-9606.
- Dyre, Jeppe C.; Hechsher, Tina; Niss, Kristine (2009). "A brief critique of the Adam–Gibbs entropy model". Journal of Non-Crystalline Solids. 355 (10–12): 624–627. arXiv:0901.2104. Bibcode:2009JNCS..355..624D. doi:10.1016/j.jnoncrysol.2009.01.039. S2CID 53051058.
- Hecksher, Tina; Nielsen, Albena I.; Olsen, Niels Boye; Dyre, Jeppe C. (2008). "Little evidence for dynamic divergences in ultraviscous molecular liquids". Nature Physics. 4 (9): 737–741. Bibcode:2008NatPh...4..673H. doi:10.1038/nphys1033. ISSN 1745-2473. Archived from the original on 2021-11-04. Retrieved 2020-08-02.
- Ojovan M.I. (2008). "Configurons: thermodynamic parameters and symmetry changes at glass transition" (PDF). Entropy. 10 (3): 334–364. Bibcode:2008Entrp..10..334O. doi:10.3390/e10030334. Archived (PDF) from the original on 2009-07-11. Retrieved 2009-09-25.
- Ojovan, M.I. (2008). "Configurons: thermodynamic parameters and symmetry changes at glass transition" (PDF). Entropy. 10 (3): 334–364. Bibcode:2008Entrp..10..334O. doi:10.3390/e10030334. Archived (PDF) from the original on 2009-07-11. Retrieved 2009-09-25.
- Ojovan, Michael I; Travis, Karl P; Hand, Russell J (2007). "Thermodynamic parameters of bonds in glassy materials from viscosity–temperature relationships" (PDF). Journal of Physics: Condensed Matter. 19 (41): 415107. Bibcode:2007JPCM...19O5107O. doi:10.1088/0953-8984/19/41/415107. PMID 28192319. S2CID 24724512. Archived (PDF) from the original on 2018-07-25. Retrieved 2019-07-06.
- Cowie, J. M. G. and Arrighi, V., Polymers: Chemistry and Physics of Modern Materials, 3rd Edn. (CRC Press, 2007) ISBN 0748740732
- Zaccone, A.; Terentjev, E. (2013). "Disorder-Assisted Melting and the Glass Transition in Amorphous Solids". Physical Review Letters. 110 (17): 178002. arXiv:1212.2020. Bibcode:2013PhRvL.110q8002Z. doi:10.1103/PhysRevLett.110.178002. PMID 23679782. S2CID 15600577.
- Capponi, S.; Alvarez, F.; Racko, D. (2020), "Free Volume in a PVME Polymer–Water Solution", Macromolecules, 53 (12): 4770–4782, Bibcode:2020MaMol..53.4770C, doi:10.1021/acs.macromol.0c00472, hdl:10261/218380, S2CID 219911779
- Slater, J.C., Introduction to Chemical Physics (3rd Ed., Martindell Press, 2007) ISBN 1178626598
- Born, Max (2008). "On the stability of crystal lattices. I". Mathematical Proceedings of the Cambridge Philosophical Society. 36 (2): 160–172. Bibcode:1940PCPS...36..160B. doi:10.1017/S0305004100017138. S2CID 104272002.
- Born, Max (1939). "Thermodynamics of Crystals and Melting". The Journal of Chemical Physics. 7 (8): 591–603. Bibcode:1939JChPh...7..591B. doi:10.1063/1.1750497.
- Frenkel, J. (1946). Kinetic Theory of Liquids. Clarendon Press, Oxford.
- Bartenev, G. M., Structure and Mechanical Properties of Inorganic Glasses (Wolters – Noordhoof, 1970) ISBN 9001054501
- Dyre, Jeppe C.; Olsen, Niels Boye; Christensen, Tage (1996). "Local elastic expansion model for viscous-flow activation energies of glass-forming molecular liquids". Physical Review B. 53 (5): 2171–2174. Bibcode:1996PhRvB..53.2171D. doi:10.1103/PhysRevB.53.2171. ISSN 0163-1829. PMID 9983702.
- Reynolds, C. L. Jr. (1979). "Correlation between the low temperature phonon mean free path and glass transition temperature in amorphous solids". J. Non-Cryst. Solids. 30 (3): 371. Bibcode:1979JNCS...30..371R. doi:10.1016/0022-3093(79)90174-1.
- Rosenburg, H. M. (1963) Low Temperature Solid State Physics. Clarendon Press, Oxford.
- Kittel, C. (1946). "Ultrasonic Propagation in Liquids". J. Chem. Phys. 14 (10): 614. Bibcode:1946JChPh..14..614K. doi:10.1063/1.1724073. hdl:1721.1/5041.
- Kittel, C. (1949). "Interpretation of the Thermal Conductivity of Glasses". Phys. Rev. 75 (6): 972. Bibcode:1949PhRv...75..972K. doi:10.1103/PhysRev.75.972.
- Chen, Shao-Ping; Egami, T.; Vitek, V. (1985). "Orientational ordering of local shear stresses in liquids: A phase transition?". Journal of Non-Crystalline Solids. 75 (1–3): 449. Bibcode:1985JNCS...75..449C. doi:10.1016/0022-3093(85)90256-X.
- Sorkin, Viacheslav (2005). "Lindemann criterion" (PDF). Point Defects, Lattice Structure and Melting (MSc). Israel Institute of Technology. pp. 5–8. Archived (PDF) from the original on 19 August 2019. Retrieved 6 January 2022.
- Mott, N. F. (1934). "The Resistance of Liquid Metals". Proceedings of the Royal Society A. 146 (857): 465. Bibcode:1934RSPSA.146..465M. doi:10.1098/rspa.1934.0166.
- Lindemann, C. (1911). "On the calculation of molecular natural frequencies". Phys. Z. 11: 609.
- Klement, W.; Willens, R. H.; Duwez, POL (1960). "Non-crystalline Structure in Solidified Gold–Silicon Alloys". Nature. 187 (4740): 869. Bibcode:1960Natur.187..869K. doi:10.1038/187869b0. S2CID 4203025.
- Duwez, Pol; Willens, R. H.; Klement, W. (1960). "Continuous Series of Metastable Solid Solutions in Silver-Copper Alloys" (PDF). Journal of Applied Physics. 31 (6): 1136. Bibcode:1960JAP....31.1136D. doi:10.1063/1.1735777. Archived (PDF) from the original on 2017-12-02. Retrieved 2018-05-16.
- Duwez, Pol; Willens, R. H.; Klement, W. (1960). "Metastable Electron Compound in Ag-Ge Alloys" (PDF). Journal of Applied Physics. 31 (6): 1137. Bibcode:1960JAP....31.1137D. doi:10.1063/1.1735778. Archived (PDF) from the original on 2020-04-18. Retrieved 2019-07-06.
- Chaudhari, P; Turnbull, D (1978). "Structure and properties of metallic glasses". Science. 199 (4324): 11–21. Bibcode:1978Sci...199...11C. doi:10.1126/science.199.4324.11. PMID 17841932. S2CID 7786426.
- Chen, J. S. (1980). "Glassy metals". Reports on Progress in Physics. 43 (4): 353. Bibcode:1980RPPh...43..353C. doi:10.1088/0034-4885/43/4/001. S2CID 250804009.
- Jonson, M.; Girvin, S. M. (1979). "Electron-Phonon Dynamics and Transport Anomalies in Random Metal Alloys". Phys. Rev. Lett. 43 (19): 1447. Bibcode:1979PhRvL..43.1447J. doi:10.1103/PhysRevLett.43.1447.
- Turnbull, D. (1974). "Amorphous Solid Formation and Interstitial Solution Behavior in Metallic Alloy System". J. Phys. C. 35 (C4): C4–1. CiteSeerX 10.1.1.596.7462. doi:10.1051/jphyscol:1974401. S2CID 52102270.
- Chen, H. S.; Park, B. K. (1973). "Role of chemical bonding in metallic glasses". Acta Metall. 21 (4): 395. doi:10.1016/0001-6160(73)90196-X.
- Wang, R.; Merz, D. (1977). "Polymorphic bonding and thermal stability of elemental noncrystalline solids". Physica Status Solidi A. 39 (2): 697. Bibcode:1977PSSAR..39..697W. doi:10.1002/pssa.2210390240.
External links
- Fragility Archived 2007-06-28 at the Wayback Machine
- VFT Eqn.
- Polymers I
- Polymers II
- Angell: Aqueous media
- DoITPoMS Teaching and Learning Package- "The Glass Transition in Polymers"
- Glass Transition Temperature short overview