Goodpasture syndrome
Goodpasture syndrome (GPS), also known as anti–glomerular basement membrane disease, is a rare autoimmune disease in which antibodies attack the basement membrane in lungs and kidneys, leading to bleeding from the lungs, glomerulonephritis,[1] and kidney failure.[2] It is thought to attack the alpha-3 subunit of type IV collagen, which has therefore been referred to as Goodpasture's antigen.[3] Goodpasture syndrome may quickly result in permanent lung and kidney damage, often leading to death. It is treated with medications that suppress the immune system such as corticosteroids and cyclophosphamide, and with plasmapheresis, in which the antibodies are removed from the blood.
| Goodpasture syndrome | |
|---|---|
| Other names | Goodpasture's syndrome, Goodpasture disease, Goodpasture's disease, anti–glomerular basement membrane disease, anti–glomerular basement membrane antibody disease, anti-GBM disease, anti-GBM antibody disease |
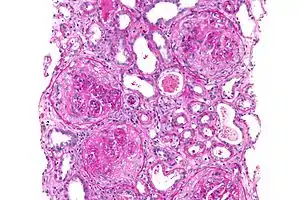 | |
| Micrograph of a crescentic glomerulonephritis that was shown to be anti–glomerular basement membrane disease, PAS stain | |
| Specialty | Nephrology, pulmonology, immunology |
The disease was first described by an American pathologist Ernest Goodpasture of Vanderbilt University in 1919 and was later named in his honor.[4][5]
Signs and symptoms
The anti–glomerular basement membrane (GBM) antibodies primarily attack the kidneys and lungs, although, generalized symptoms like malaise, weight loss, fatigue, fever, and chills are also common, as are joint aches and pains.[6] 60 to 80% of those with the condition experience both lung and kidney involvement; 20-40% have kidney involvement alone, and less than 10% have lung involvement alone.[6] Lung symptoms usually antedate kidney symptoms and usually include: coughing up blood, chest pain (in less than 50% of cases overall), cough, and shortness of breath.[7] Kidney symptoms usually include blood in the urine, protein in the urine, unexplained swelling of limbs or face, high amounts of urea in the blood, and high blood pressure.[6]
Cause
While the exact cause is unknown, the genetic predisposition to GPS involves the human leukocyte antigen (HLA) system, specifically HLA-DR15.[8] In addition to genetic susceptibility, an initial environmental insult to the pulmonary vasculature is needed to allow the anti-glomerular basement membrane (anti-GBM) antibodies to reach the alveolar capillaries. Examples of such an insult include: exposure to organic solvents (e.g. chloroform) or hydrocarbons, exposure to tobacco smoke, infection (such as influenza A), cocaine inhalation, metal dust inhalation, bacteremia, sepsis, high-oxygen environments, and antilymphocyte therapies (especially with monoclonal antibodies).[9] Exposure to dry cleaning chemicals and Paraquat brand weed killer have also been implicated as potential insults. [10] In GPS, anti-GBM antibodies are produced and circulated throughout the bloodstream, damaging the membranes lining the lungs and kidneys as well as targeting their capillaries.[11]
Pathophysiology
GPS is caused by abnormal plasma cell production of anti-GBM antibodies.[9] The major target of these abnormal antibodies is the non-collagen domain of the alpha-3 chain of type 4 collagen, which is mostly found in the basal membranes of glomerular and alveolar capillaries, explaining the obscurely specific symptoms of this condition.[12] This preferred targeting of these alpha-3 collagen chains specifically in the basal membranes of glomerular and alveolar capillaries can be explained by the higher accessible exposure of epitopes, a larger expansion of the alpha-3 collagen units, and because these alpha-3 collagen chains structurally provide higher accessibility for the targeting antibodies. [13] These antibodies bind their reactive epitopes to the basement membranes and activate the complement cascade, leading to the death of tagged cells.[9] A specific antibody and epitope binding that shows the highest affinity and is pathogenic occurs between GPA antibodies and the anti-GBM epitope region, designated EA, which is residues 17-31 of the alpha 3 subunit of non-collagenous domain of type IV collagen.[14] T cells are also implicated, though it is generally considered a type II hypersensitivity reaction.[9]
Diagnosis
The diagnosis of GPS is often difficult, as numerous other diseases can cause the various manifestations of the condition and the condition itself is rare.[15] The most accurate means of achieving the diagnosis is testing the affected tissues by means of a biopsy, especially the kidney, as it is the best-studied organ for obtaining a sample for the presence of anti-GBM antibodies.[15] On top of the anti-GBM antibodies implicated in the disease, about one in three of those affected also has cytoplasmic antineutrophilic antibodies in their bloodstream, which often predates the anti-GBM antibodies by about a few months or even years.[15] The later the disease is diagnosed, the worse the outcome is for the affected person.[9]
In addition, if there is substantial suspicion of the disease, seralogic testing for ELISA assay is usually done by looking for alpha3 NC1 domain area of collagen IV in order to avoid false positives.[16]
Treatment
The major mainstay of treatment for GPS is plasmapheresis, a procedure in which the affected person's blood is sent through a centrifuge and the various components separated based on weight.[17] The plasma contains the anti-GBM antibodies that attack the affected person's lungs and kidneys, and is filtered out.[17] The other parts of the blood (the red blood cells, white blood cells, and platelets) are recycled and intravenously reinfused.[17] Most individuals affected by the disease also need to be treated with immunosuppressant drugs, especially cyclophosphamide, prednisone, and rituximab, to prevent the formation of new anti-GBM antibodies so as to prevent further damage to the kidneys and lungs.[17] Other, less toxic immunosuppressants such as azathioprine may be used to maintain remission.[17]
Prognosis
With treatment, the five-year survival rate is >80% and fewer than 30% of affected individuals require long-term dialysis.[9] A study performed in Australia and New Zealand demonstrated that in patients requiring renal replacement therapy (including dialysis) the median survival time is 5.93 years.[9] Without treatment, virtually every affected person will die from either advanced kidney failure or lung hemorrhages.[9]
Epidemiology
GPS is rare, affecting about 0.5–1.8 per million people per year in Europe and Asia.[9] It is also unusual among autoimmune diseases in that it is more common in males than in females and is also less common in blacks than whites, but more common in the Māori people of New Zealand.[9] The peak age ranges for the onset of the disease are 20–30 and 60–70 years.[9]
See also
- HLA-DR § DR2
- Pulmonary-renal syndrome
References
- "Goodpasture Syndrome". www.hopkinsmedicine.org. Retrieved 2020-12-05.
- Thibaud, V.; Rioux-Leclercq, N.; Vigneau, C.; Morice, S. (December 2019). "Recurrence of Goodpasture syndrome without circulating anti-glomerular basement membrane antibodies after kidney transplant, a case report". BMC Nephrology. 20 (1): 6. doi:10.1186/s12882-018-1197-6. ISSN 1471-2369. PMC 6323659. PMID 30621605.
- "COL4A3 gene".
- Goodpasture EW (1919). "The significance of certain pulmonary lesions in relation to the etiology of influenza". Am J Med Sci. 158 (6): 863–870. doi:10.1097/00000441-191911000-00012. S2CID 71773779.
- Salama AD, Levy JB, Lightstone L, Pusey CD (September 2001). "Goodpasture's disease". Lancet. 358 (9285): 917–920. doi:10.1016/S0140-6736(01)06077-9. PMID 11567730. S2CID 40175400.
- Kathuria, P; Sanghera, P; Stevenson, FT; Sharma, S; Lederer, E; Lohr, JW; Talavera, F; Verrelli, M (21 May 2013). Batuman, C (ed.). "Goodpasture Syndrome Clinical Presentation". Medscape Reference. WebMD. Retrieved 14 March 2014.
- Schwarz, MI (November 2013). "Goodpasture Syndrome: Diffuse Alveolar Hemorrhage and Pulmonary-Renal Syndrome". Merck Manual Professional. Retrieved 14 March 2014.
- "Goodpasture syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2020-12-01.
- Kathuria, P; Sanghera, P; Stevenson, FT; Sharma, S; Lederer, E; Lohr, JW; Talavera, F; Verrelli, M (21 May 2013). Batuman, C (ed.). "Goodpasture Syndrome". Medscape Reference. WebMD. Retrieved 14 March 2014.
- "Goodpasture Syndrome".
- "Goodpasture Syndrome". NORD (National Organization for Rare Disorders). Retrieved 2020-11-29.
- Marques, C., et al. (2020). Review on anti-glomerular basement membrane disease or Goodpasture's syndrome. The Journal of Internal Medicine, 41(1), 14-20.
- Greco, A., et al. (2015). Goodpasture's syndrome: A clinical update. Autoimmunity Reviews, 14 (3), 246-253.
- Borza, D., et al. (2003). Pathogenesis of Goodpasture syndrome: a molecular perspective. Seminars in Nephrology, 23(6), 522-531.
- Kathuria, P; Sanghera, P; Stevenson, FT; Sharma, S; Lederer, E; Lohr, JW; Talavera, F; Verrelli, M (21 May 2013). Batuman, C (ed.). "Goodpasture Syndrome Workup". Medscape Reference. WebMD. Retrieved 14 March 2014.
- "Goodpasture Syndrome". NCBI. 25 March 2020. Retrieved 20 December 2020.
- Kathuria, P; Sanghera, P; Stevenson, FT; Sharma, S; Lederer, E; Lohr, JW; Talavera, F; Verrelli, M (21 May 2013). Batuman, C (ed.). "Goodpasture Syndrome Treatment & Management". Medscape Reference. WebMD. Retrieved 14 March 2014.