Pheochromocytoma
Pheochromocytoma (PHEO or PCC) is a rare tumor of the adrenal medulla composed of chromaffin cells, also known as pheochromocytes.[3] When a tumor composed of the same cells as a pheochromocytoma develops outside the adrenal gland, it is referred to as a paraganglioma.[4] These neuroendocrine tumors are capable of producing and releasing massive amounts of catecholamines, metanephrines, or methoxytyramine, which result in the most common symptoms, including hypertension (high blood pressure), tachycardia (fast heart rate), and diaphoresis (sweating).[5] However, not all of these tumors will secrete catecholamines. Those that do not are referred to as biochemically silent, and are predominantly located in the head and neck.[6] While patients with biochemically silent disease will not develop the typical disease manifestations described above, the tumors grow and compress the surrounding structures of the head and neck, and can result in pulsatile tinnitus (ringing of the ear), hearing loss, aural fullness, dyspnea (difficulty breathing), and hoarseness.[7] While tumors of the head and neck are parasympathetic, their sympathetic counterparts are predominantly located in the abdomen and pelvis, particularly concentrated at the organ of Zuckerkandl.[8]
| Pheochromocytoma | |
|---|---|
| Other names | Phaeochromocytoma, adrenal medullary tumor, Chromaffin Cell Tumors, Paraganglioma |
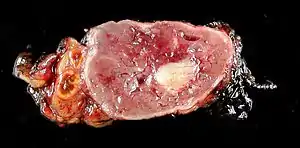 | |
| Normal remnant adrenal gland (left) with a pheochromocytoma (right) involving the adrenal medulla | |
| Pronunciation |
|
| Specialty | Endocrinology, oncology |
| Symptoms | Hypertension, tachycardia, sweating, headache, pallor |
| Complications | Hypertensive crisis |
| Diagnostic method | Elevated plasma free metanephrines, plasma catecholamines, or urinary catecholamines |
| Treatment | Surgery, chemotherapy, radiation, and pharmacologic agents |
| Frequency | 0.8 per 100,000 person-years [2] |
Signs and symptoms
The signs and symptoms of a pheochromocytoma are those related to sympathetic nervous system hyperactivity.[9] The classic triad includes headaches (likely related to elevated blood pressure, or hypertension), tachycardia/elevated heart rate, and diaphoresis (excessive sweating, particularly at night, also known as hyperhidrosis). However, patients are unlikely to experience continuous symptoms. Due to the paroxysmal nature of catecholamine synthesis and release, patients may experience "attacks" or "spells" where they are suddenly overwhelmed with signs and symptoms of their tumor.[10] Attacks can occur spontaneously (without warning) or may be triggered by a variety of pharmaceutical agents, foods, intraoperative tumor manipulation, intubation, or during anesthetic induction.[11]
| Lifestyle | Medications | Diet |
|---|---|---|
| Physical Exertion | Histamine | Cheese |
| Anxiety/Stress | Metoclopramide | Fermented wine/beer |
| Trauma/Pain | Glucagon | Tomatoes |
| Micturition | ACTH | Coffee/Beans |
While the above symptoms are classic, other common clinical manifestations have been reported and include (in no particular order)[5][11]
- Pallor
- Heat intolerance
- Weight loss
- Chest and/or Abdominal Discomfort
- Nausea/Vomiting
- Constipation
- Orthostatic Hypotension
- Medically defined as a decrease in systolic blood pressure (top number) of 20 mm Hg or diastolic blood pressure (bottom number) of 10 mm Hg after a change in position from lying down or sitting to a standing position[13]
- Feeling of becoming light-headed or dizzy after swiftly changing positions
- Psychiatric Manifestations
- Anxiety, Panic Attacks, Nervousness, Tremulousness
- Hyperglycemia (high blood sugar)
Complications
While the symptoms of a pheochromocytoma are quite common, the disease has been referred to as "the great mimic".[14] Literature reports that just 0.1% of patients with hypertension are diagnosed with this rare endocrine disorder and symptomatic patients are often mistaken for much more common diseases.[15] As symptoms are often paroxysmal (episodic/sporadic), patients may not immediately seek treatment as the problem "disappears on its own." Furthermore, when pictured in the ideal clinical scenario (an older woman in her mid-50s), the spontaneous attacks of flushing, sweating, and a racing heart may be mistaken for pre-menopausal related hot-flashes. Unmanaged pheochromocytoma is dangerous and can lead to serious complications, including death.[16][17] The cardiovascular system is the most commonly involved.[18][19][20]
In pregnancy, pheochromocytoma is associated with significant maternal and fetal mortality, mainly due to hypertensive crisis in the mother and intrauterine growth restriction in the fetus.[21][22]
Cardiovascular system
- Hypertensive crisis: Pheochromocytoma-related hypertensive emergencies are one of the most feared clinical manifestations. Attacks are random and may occur secondary to a trigger (see Signs and Symptoms above) or spontaneously after a catecholamine surge.[19] The prevailing symptom is elevated systolic blood pressure (> 200 mmHg) that is unresponsive to traditional treatment regimens and threatens end-organ damage.[18] Patients require immediate, life-saving treatment to prevent further damage to other organs and/or death.
- Myocardial Ischemia/Infarction: A heart attack is often caused by a significant build-up of plaque (atherosclerosis) in the coronary vessels. Patients with pheochromocytoma present with myocardial infarctions despite an overall lack of plaque build-up, indicating a different mechanism for the myocardial infarction. Current research hypothesizes that the tumor secretes massive amounts of catecholamines, which directly interact with myocardial (heart) tissue and exert negative effects including oxygen deprivation, leading to accelerated scarring and cell death.[18]
- Toxic Myocarditis: Even in patients without myocardial damage, excessive catecholamines can result in abnormal ST changes on an ECG. Norepinephrine (a catecholamine) is hypothesized to result in damaged cardiac tissue by inhibiting coronary blood flow and depriving cells of oxygen, thus resulting in ischemic tissue.[20] Fortunately, following tumor excision and the subsequent quelling of catecholamines, the damage has been proven reversible.
- Cardiomyopathy: Pheochromocytomas have been implicated in various types of cardiomyopathy, including (myocarditis, see above), dilated cardiomyopathy, and stress-induced or Takotsubo cardiomyopathy.[23] As with the other cardiovascular-related complications, excess catecholamines are responsible for the increased myocardial burden and significant physiologic stress.[24] Current literature indicates that most of the catecholamine-induced damage is reversible, thereby strengthening the argument for early and accurate diagnosis in order to allow for cardiac remodeling and prevent further destruction.[23][24]
- Arrhythmias: Sinus tachycardia is the most common abnormal heart rhythm associated with a pheochromocytoma and is experienced by patients as the feeling of a "fluttering heart" or palpitations.[18] Many other tachyarrhythmias (fast heart rate) have also been reported.
Nervous system
- Cerebrovascular Accident (Stroke): Multiple reports have detailed transient ischemic attacks or strokes in patients with a pheochromocytoma.[25][26][27][28][29][30][31] In a study of 130 patients with pheochromocytoma, 7 patients were diagnosed with a transient ischemic attack (the neurologic deficit completely resolved) and 3 patients experienced a stroke with persistent symptoms.[32]
- Headache: Headaches are one of the core clinical manifestations of a pheochromocytoma and can result in debilitating pain. The majority of studied patients report their pain began and ended abruptly without warning and described the pain as a severe, bilateral throbbing (although the scale of severity was not published). While 71% of the studied patients reported headaches, just over 20% of the affected patients endorsed associated nausea, vomiting, photophobia, or phonophobia, which are typically associated with migraines.[33]
Urinary system
- Acute Renal Failure: Several reports have detailed rhabdomyolysis (rapid skeletal muscle breakdown) leading to acute kidney injury and the need for transient dialysis in the undiagnosed pheochromocytoma patient as their primary presenting symptom.[34][35][36][37] Kidney failure is brought about by catecholamine-induced muscle injury. Norepinephrine causes vessels to narrow, thereby limiting blood flow and inducing ischemia.[34]
Multiple organ dysfunction syndrome (MODS)[38]: Caused by an elevated inflammatory response, multiple organ dysfunction is a severe, life-threatening emergency with increasing mortality based on the number of systems involved.[39] Pheochromocytoma-related MODS is associated with multiple organ failure, hyperthermia > 40 degrees Celsius, neurologic manifestations, and cardiovascular instability resulting in either hypo or hypertension.[40] In contrast to a hypertensive crisis, pheochromocytoma-associated MODS may not respond to traditional alpha-receptor agents and may require emergent surgical excision if clinical stability is not achieved.
Genetics
Current estimates predict that upwards of 40% of all pheochromocytomas are related to an inherited germline susceptibility mutation.[41] Of the remaining 60% of tumors, more than 30% are associated with a somatic mutation.[42] Given the high association with genetic inheritance, the United States Endocrine Society recommends that all patients diagnosed with a pheochromocytoma undergo an evaluation with a genetic counselor to consider genetic testing.[43] The most recent data indicates that there are 25 pheochromocytoma susceptibility genes; however, just 12 are recognized as part of a well-known syndrome.[8] Determining the genetic status of a pheochromocytoma patient is crucial – each gene is inherited in a different pattern, associated with specific disease characteristics, and may respond more favorably to certain treatment options. Furthermore, early identification can guide physicians on screening recommendations for first degree relatives of patients with pheochromocytoma.[44] There is no current consensus for how and when asymptomatic carriers (individual who has a genetic variant associated with pheochromocytoma, but no current evidence of disease) should be evaluated. Conversations should occur at an individual level with the patient and their provider to develop a personalized screening plan that alternates between a biochemical (blood work) evaluation and whole-body imaging to monitor disease progression.[45]
Pediatric considerations
Additional practices may help maintain the emotional and psychological well-being of the minor. Screening includes a multidisciplinary team (endocrinologist, oncologist, psychologist, geneticist, parent, and child) where the primary focus is supporting the child.[46]
- A positive result from testing during family-observed days of celebration may mask the happiness associated with these events in the future.
- Testing one pediatric sibling at a time allows the family to narrow their focus when results are returned and support each sibling individually.
- A negative result may be upsetting to a child if their sibling was positive; an opportunity to ask questions and process results may be helpful.
Hereditary syndromes
The following table(s) detail the clinical characteristics of the well-known hereditary pheochromocytoma gene variants[47][48][49][44][42][41][50]
| Gene | Inheritance | Penetrance | Metastatic Potential | 1o Disease Characteristics | |
|---|---|---|---|---|---|
| MEN2 | RET | Autosomal Dominant | 40–50% | <5% | Medullary thyroid carcinoma, hyperparathyroidism, marfanoid habitus, pheochromocytoma |
| VHL | VHL | 10-30% | 5% | Renal cell carcinoma, pancreatic NET, retinal and CNS hemangioblastoma, pheochromocytoma | |
| NF1 | NF1 | 1–5% | 12% | Neurofibromas, cafe-au-lait macules, lisch nodules,pheochromocytoma |
MEN2 (Multiple Endocrine Neoplasia-2); VHL (von-Hippel Lindau); NF1 (Neurofibromatosis-1); NET (Neuroendocrine Tumor); CNS (Central Nervous System)
| Gene | Inheritance | Penetrance | Metastatic Potential | 1o Disease Characteristics | |
|---|---|---|---|---|---|
| PGL1 | SDHD | Autosomal Dominant
Paternal Inheritance |
90% | <5% | Head and neck paraganglioma, pheochromocytoma, gastrointestinal stromal tumor |
| PGL2 | SDHAF2 | 100% | Low | Head and neck paraganglioma | |
| PGL3 | SDHC | Autosomal Dominant | Inconsistent | Inconsistent | Pheochromocytoma, head and neck paraganglioma, gastrointestinal stromal tumor |
| PGL4 | SDHB | 30–50% | 30–70% | Head and neck paraganglioma, pheochromocytoma, gastrointestinal stromal tumor | |
| PGL5 | SDHA | 10–15% | Low | Pheochromocytoma, head and neck paraganglioma, gastrointestinal stromal tumor |
SDHx (Succinate Dehydrogenase Subunit x)
| Inheritance | Penetrance | Metastatic Potential | 1o Disease Characteristics | |
|---|---|---|---|---|
| MAX | Autosomal Dominant | Inconsistent | <5% | Bilateral pheochromocytoma |
| TMEM127 | Inconsistent | Low | Pheochromocytoma, head and neck paraganglioma |
MAX (MYC Associated Factor X); TMEM127 (Transmembrane Protein 127)
Other gene variants
There have been several published case reports of other, rare pheochromocytoma-associated susceptibility genes:
- Pacak-Zhuang Syndrome[51][52][53][54][55]
- Hypoxia-inducible factor 2 alpha (HIF2A)
- Polycythemia
- Duodenal somatostatinoma
- Retinal and choroidal vascular changes
- Paraganglioma/Pheochromocytoma
- Pheochromocytoma and Giant Cell Tumor of Bone[56]
- H3 histone, family 3A (H3F3A), post-zygotic G34W
- Pheochromocytoma/Paraganglioma
- Carney Triad[57]
- Gastrointestinal stromal tumor
- Pulmonary chondroma
- Paraganglioma
- Carney-Stratakis Syndrome[58]
- Gastrointestinal stromal tumor
- Paraganglioma
Several additional gene variants have been described, but the provided information is inconsistent and a consensus has not been reached in the community if these mutations are truly pheochromocytoma susceptibility genes.
Diagnosis
Differential
If a patient has the characteristic signs and symptoms of a pheochromocytoma and the decision is made to pursue additional biochemical (blood work) evaluation, the differential diagnosis is important as it is more likely to be something other than a pheochromocytoma given the relative frequency of 0.8 per 100,000 person-years.[2]
| Endocrine | Cardiovascular | Neurologic | Psychiatric | Other |
|---|---|---|---|---|
| Hyperthyroidism | Heart Failure | Migraine | Anxiety | Porphyria |
| Carcinoid Syndrome | Arrhythmias | Stroke | Panic Disorder | Medications[lower-alpha 2] |
| Hypoglycemia | Ischemic Heart Disease | Epilepsy | Substance Use[lower-alpha 3] | |
| Menopausal Syndrome | Baroreflex Failure | Meningioma | Factitious Disorder[lower-alpha 4] | |
| Medullary Thyroid Carcinoma | – | POTS | – |
Notes
- Adopted from Lenders et al., Phaeochromocytoma. The Lancet. 366(9486); 665–675.[3]
- Monoamine Oxidase Inhibitors, Clonidine Withdrawal
- Including but not limited to cocaine use
- Misuse of over-the-counter medications such as pseudoephedrine hat are sympathomimetics
Gold standard
Elevated plasma free metanephrines is considered the gold standard diagnosis for pheochromocytoma.[59] Over 10 studies have confirmed that the sensitivity and specificity of this test is 97% and 93% respectively; however, there is still concern for false positive results in the correct clinical scenario.[5] When interpreting a biochemical analysis for pheochromocytoma, the provider must pay close attention to the (1) conditions of the collection, (2) all medications the patient is taking, and (3) their diet.[60]
- Conditions of Collection: Unlike many routine laboratory tests that can be drawn at a moments notice, there are several recommendations that should be followed to ensure the ideal conditions and an accurate sample. Current research indicates that blood work should only be drawn after a patient has been resting supine (flat on their back) for 30 minutes before collection.[61][62] Specific supine reference values should be used in this scenario. Ensuring these conditions is difficult and may be cost-prohibitive at most institutions. In these cases, a rested, supine draw can be repeated following a positive result in a seated position to eliminate false-positive results.[60]
- Pharmaceutical Interference: Many prescription, over-the-counter, and illicit substances can interfere with the proper collection of plasma metanephrines and lead to false-positive results. Providers should review a patient's medication list in-detail and have a discussion if temporarily discontinuing any of the interfering medications is possible. The most reported medications to result in falsely elevated metanephrines include: β-adrenoceptor blockers, phenoxybenzamine, tricyclic antidepressants, monoamine oxidase inhibitors, serotonin norepinephrine reuptake inhibitors (SNRI), and methyldopa.[63][60] As the majority of these medications are commonly prescribed for psychiatric conditions, a conversation with the prescriber may be necessary to facilitate alternative therapeutic options while the patient is undergoing evaluation for a pheochromocytoma.[63] After any possible prescription medications have been held, it is important to review any over-the-counter medications/supplements as well as the commonly used acetaminophen and pseudoephedrine cause false elevations in metanephrine levels.[60][63] Finally, it is important to have open, non-judgemental discussions about the patient's recreational substance use. Amphetamines, nicotine, and cocaine can result in marked plasma norepinephrine levels.
- Lifestyle and Diet: As with most lab work, the patient should refrain from eating (fasting) after midnight the night prior to their collection. However, there are further recommendations specific to a metanephrines collection, including abstaining from nicotine, alcohol, and exercise for at least 12 hours prior to their lab draw.[8] Patients should also avoid catecholamine-containing foods (fruits, fruit drinks, chocolate, caffeine, tomatoes, beans, nuts, and potatoes) for a minimum of 24 hours prior to collection.[64][65]
While the above (3) conditions are likely to contribute to false-positive results if not controlled for, any value greater than 3 to 4 times the upper reference limit of normal should be considered diagnostic for a pheochromocytoma.[43][66]
Alternative tests
Twenty-four hour urinary metanephrines are an acceptable alternative if the plasma test is unavailable.[67] Other additional biomarkers can be helpful to aid in the diagnosis of pheochromocytoma as well, most notable is Chromogranin A. In comparison to the specificity of elevated catecholamines in the pheochromocytoma patient, chromogranin A is a non-specific polypeptide that is high in a variety of neuroendocrine tumors.[68] However, a 2006 report from Italy found that over 90% of studied pheochromocytoma patients demonstrated elevated chromogranin A levels.[69] If metanephrine values are equivocal, chromogranin A can be used as an adjunct marker to predict the presence of a tumor.
Borderline elevated metanephrines present a diagnostic challenge to the physician - the first step is to repeat the labs, taking extra precautions to follow the gold standard diagnosis described above, including the conditions of collection, pharmaceutical interference, and any potential diet and lifestyle habits that could alter the results. If the offending medications cannot be discontinued or repeated labs remained the same, consider administering a clonidine suppression test.[70] In the 1970s, the drug clonidine hydrocloride swept the market as a novel agent for hypertension; however, the reported side-effects (nausea, vomiting, drowsiness, dryness of the eyes and mouth, constipation, and generalized weakness) limit compliance and have vastly diminished prescriptions.[71] While the adverse side-effects with clonidine are inconvenient, the most dangerous aspect of clonidine is withdrawal rebound hypertension - that is, when the medicine is abruptly discontinued, blood pressure may rapidly return or surpass the original value.[72][73][74] However, a one-time, weight-based dose can be utilized in limited settings to help determine disease status.[60] After fasting overnight, patient's will present to their testing site for a baseline metanephrines blood draw and clonidine administration. They will remain supine for (3) hours and a repeat blood draw will be taken. A positive result (indicating a pheochromocytoma) will occur if the plasma metanephrine levels remain elevated after clonidine is given. If the results are the same or fall, the test is negative and the patient does not have a pheochromocytoma.[60] It is important to note that if a patient does not have a pheochromocytoma, they may become extremely hypotensive following clonidine. Patients should not depend on themselves for transport following this test.
Plasma methoxytyramine is a breakdown product of the catecholamine, dopamine. Paragangliomas of the head and neck commonly secrete dopamine, but are referred to as "biochemically silent" because they do not cause the characteristic symptoms associated with a pheochromocytoma. However, methoxytyramine can be utilized to detect the tumors of the head and neck.[75] Further research indicates that the biomarker is also a useful indicator of metastatic disease - which is the only current biochemical evidence of metastases to date.[76]
Biochemical phenotypes
While diagnostic, laboratory values can also provide physician's with important information about the type, location, size, and associated tumor genotype.[66] There are (3) major, well-recognized biochemical phenotypes that can be used by health care providers to direct patient care.[77]
- Adrenergic (Epinephrine and metanephrine)
- More likely to indicate an adrenal tumor[78]
- When plasma metanephrine levels were elevated to greater than 15% of the combined levels of normetanephrine and metanephrine, an adrenal tumor or a recurrence of an adrenal tumor that had already been excised can be predicted
- Patients are more likely to present with the classic, paroxysmal (episodic) symptoms described above[66]
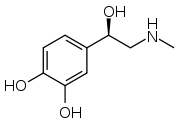
- Noradrengeric (Norepinephrine and normetanephrine)
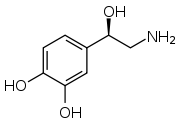 Structure of norepinephrine
Structure of norepinephrine- More likely to indicate an extra-adrenal tumor[78]
- Patients are more likely to present with continuous, persistent pheochromocytoma-related symptoms (hypertension and tachycardia) compared to those that are classically episode with an adrenergic phenotype[66]
- Common in patients with von-Hippel Lindau and succinate dehydrogenase subunit X genetic variants[66]
- Dopaminergic (Dopamine and 3-methoxytyramine)
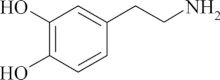 Structure of dopamine
Structure of dopamine- More likely to indicate an extra-adrenal tumor of the head and neck[77]
- Patients are more likely to be asymptomatic; however, they may present with non-specific signs of nausea, vomiting, abdominal pain, diarrhea, and weight loss secondary to the stimulation of dopamine receptors throughout the gastrointestinal tract[66]
- Particularly prevalent in patients with succinate dehydrogenase subunit B genetic variants [66]
Across both an adrenergic and a noradrenergic phenotype, the greater the sum of plasma or urinary concentrations of metanephrine and normetanephrine, the larger the expected tumor diameter.[78]
Anatomic imaging
Anatomic imaging refers to computed tomography (CT) [CAT scan] or magnetic resonance imaging (MR) scans. These imaging modalities serve to initially locate the tumor and provide detailed information about size, morphology, and structural relation to adjacent internal structures.[79] Traditionally, a patient presents to their physician for symptoms concerning for a pheochromocytoma, which prompts a biochemical evaluation. If the results are positive, the patient is referred for anatomic imaging with a CT or MR scan. However, as anatomic imaging becomes more readily available, patients are referred to an endocrinologist after an incidental (unanticipated finding) adrenal nodule is found on a scan ordered for another reason.[80] For example, "Patient M" presents to his local emergency room for abdominal pain and a CT is ordered to rule-out appendicitis; however, the radiologist notes there is a 3.5 centimeter right adrenal mass.
While there has not been a consensus on if CT or MR is the preferred imaging modality in pheochromocytoma, each method has its associated strengths and weaknesses. As CT expose the patient to ionizing radiation, MR is preferred in children and pregnant women.[81] Furthermore, the intravenous contrast used in CT can cause kidney damage and should therefore be avoided in patients with pre-existing damage.[82] However, patients who struggle with being in confined spaces for extended periods of time (claustrophobia) cannot often tolerate an MR as the machine is close-ended compared to the open-ended design of a CT.[83] When patients become anxious and begin to move in the machine, this causes motion artifact, which occurs less in CT-based images.[84]
Compared to CT and MR, ultrasound is not a preferred imaging modality and should be avoided in the pheochromocytoma patient. However, in specific patient populations where avoid ionizing radiation is the top priority (children, pregnant women), ultrasound can be used as an adjunct method when MR may be unavailable or the patient is unable to complete the scan. Furthermore, if an acute adrenal hemorrhage is suspected in a pheochromocytoma patient, ultrasound is a quick, painless, radiation-less, and cheap modality for a "first-pass" before the above imaging modalities or surgery is used to confirm the diagnosis.[85]
Functional imaging
The imaging modalities discussed below are for tumor characterization, confirmation of metastatic disease, and treatment planning - they are not used to discern tumor location or help the surgical team prepare for excision.[86] For most pheochromocytoma patients, functional imaging will follow a CT or MR. If anatomic imaging only demonstrates an adrenal tumor without evidence of disease anywhere else in the body and the metanephrine levels are overtly elevated, functional imaging can be foregone in favor of prompt surgical excision.[81] Over the last decade, there have been five functional techniques used to evaluate the pheochromocytoma patient (1) 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET), commonly referred to as the PET scan, (2) iodine-123 meta-iodobenzylguanadine (123I-MIBG), (3) 18F-flurodihydroxyphenylalanine (18F-FDOPA),(4) 68Ga-DOTA coupled somatostatin analogs (68Ga-DOTA),(5) 11C-Hydroxy ephedrine(HED-PET). From this point forward, these imaging modalities will be referred to in their abbreviated names found in parentheses.
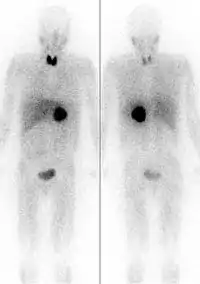
The first functional imaging technique utilized in pheochromocytoma patients was 123I-MIBG scintigraphy (Image Right). Given the compounds similar structure to the catecholamine norepinephrine (secreted by pheochromocytomas), MIBG was well-suited for uptake by most neuroendocrine tumors.[87] Furthermore, if a patient was found to be positive on an MIBG scan, they were eligible for MIBG treatment, offering additional avenues for those with widespread metastatic disease.[88] However, further investigation revealed that while MIBG excelled with adrenal lesions, it was far less superior in patients with extra-adrenal paragangliomas, particularly with specific genetic variants like succinate dehydrogenase subunit X (SDHx).[76] As the positron emission tomography scans were developed, MIBG has slowly loss its favor for the pheochromocytoma patient.[76]
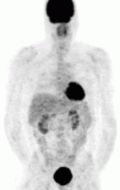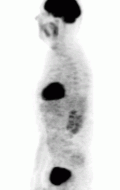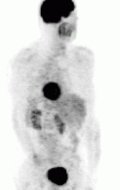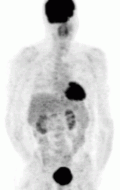
Of the four above mentioned modalities, 18F-FDG PET is the most common and readily available functional imaging technique at most hospital systems, but the least-specific to neuroendocrine tumors (Image Left). In 2012, over 200 patients participated in a trial that compared the current gold standard of the time (MIBG/CT/MRI) to the novel FDG PET. Compared to its functional counterpart, FDG outperformed MIBG in detecting soft-tissue and bone metastases with higher specificity in patients with biochemically active tumors.[76]
Following the development of FDG-PET, neuroendocrine-specific PET scans began to emerge. One of the first favorable imaging modalities was 18F-FDOPA, which demonstrated a high sensitivity in detecting head and neck paragangliomas as well as non-metastatic disease outside of the head and neck.[76][89] Unfortunately, in cases of metastatic disease, particularly related to succinate dehydrogenase subunit B (SDHB) mutations, 18F-FDOPA fell inferior to the traditional FDG-PET.[90] However, for patients with genetic variants in other pheochromocytoma-susceptibility genes (NF1, VHL, RET) 18F-FDOPA has become the preferred radiopharmaceutical agent.[91]
The newest PET modality involves somatostatin receptor type two receptor imaging with 68Ga-DOTA analogues.[84] Over the last decade, further research continues to indicate the superiority of this functional imaging modality in a wide range of clinical scenarios, even surpassing anatomic imaging (CT/MR) in pediatric patients with succinate dehydrogenase (SDHx) mutations.[92] While FDOPA inconsistently detected metastatic disease, 68Ga-DOTA analogues have demonstrated superior localization of metastatic pheochromocytoma.[93] When directly compared in one head-to-head study in 2019, 68Ga-DOTA analogues outperformed FDOPA, particularly in the detection of metastatic bone lesions.[94] An additional benefit of the DOTA analogues is the ability for treatment with peptide receptor radionuclide therapy, which will be discussed in the treatment section below.[95]
Also, HED-PET has shown to be an accurate tool to diagnose and rule out pheochromocytoma in complex clinical scenarios and to characterise equivocal adrenal tumours.[96]
Management
Surgery
Surgical resection is the only curative option for pheochromocytoma as of 2019.[97] A successful excision is a multidisciplinary effort involving the endocrinologist and the patient pre-operatively (discussed below) and the surgical team and anesthesiologist intraoperatively. Without frequent and adequate communication between all of the above-mentioned teams, a favorable outcome is much more difficult.[97] The United States Endocrine Society 2014 Clinical Practice Guideline for pheochromocytoma recommend a laparoscopic adrenalectomy (minimally invasive technique) for most adrenal tumors, unless they are invasive or are larger than 6.0 centimeters.[43] It is important to note that larger tumors can be attempted with a minimally invasive approach, but the team should be prepared to convert to an open procedure if necessary.[98] An open procedure (traditional surgical technique) is currently preferred for extra-adrenal disease, unless the tumor is small, non-invasive, and in an easy to maneuver location. While previous data indicated the need for a minimally invasive approach with malignant and/or metastatic disease, current research indicates a successful operation is feasible and results in a shorter hospital stay.[99] Literature within the last decade has also demonstrated that the robotic technique may be successfully utilized for adrenal tumors.[100]
Typically, complete or total adrenalectomy is performed; however, a technique referred to as "cortical-sparing" can leave a remnant (piece) of the adrenal gland in hopes of avoiding life-long steroid replacement if the left and right adrenal glands need to be removed.[101] The issue is particularly important in patients with MEN and VHL-related disease, which has a higher chance of bilateral pheochromocytomas.[102] The risk of leaving adrenal tissue is recurrent disease (tumor comes back). A 2019 cohort study reported that despite a 13% recurrent rate in patients who underwent a cortical-sparing adrenalectomy for pheochromocytoma, there was no decreased survival compared to their total adrenalectomy counterparts.[101]
Pre-operative management
Arguably, the most important part of a pheochromocytoma surgical plan is an adequate pre-operative blockade. Excess catecholamines have been described as a dormant volcano, ready to erupt at any time, wreaking catastrophic havoc on the body.[103] While an eruption can occur at any time, two of the most common triggers are anesthesia and direct tumor manipulation, making surgery one of the most dangerous times for a pheochromocytoma patient if not properly prepared.[104] In order to help circumvent a catecholamine-crisis, the United States Endocrine Society recommends that all patients with functional (hormonally active) tumors be started on a pre-operative alpha-adrenoceptor blockade a minimum of seven days prior to surgery.[43] There are several medication options depending on the clinical scenario, each with their own associated strengths and weaknesses.
Alpha blockade
If the patient's blood pressure is moderately elevated, a selective, short-acting alpha-1 adrenoceptor antagonist (doxazosin, prazosin, terazosin) is the preferred agent.[103] However, the patient should be warned about the potential side-effect known as "the first-dose phenomenon." When patients are initially exposed to one of the above agents, they may become lightheaded, dizzy, and nauseous, particularly when transferring from a seated to standing position due to a rapid decrease in blood pressure.[105] These effects will decrease with time, but providers can try to avoid them by starting at a low-dose and slowly increasing until they reach their desired amount. In patient's with uncontrolled hypertension, the non-selective alpha-1 and 2 adrenoceptor antagonist (phenoxybenzamine) should be utilized.[103] Unfortunately, compared to the selective agents listed above, phenoxybenzamine is much more expensive and may not be readily available to some patients. Common side-effects include dry mouth, nasal congestion, and impaired male ejaculation, all of which do not cease with time and may limit patient compliance.[106] While uncommon, patients may have a hormonally-active pheochromocytoma and a normal blood pressure. One comparison from 2014 found that a small dose of a calcium-channel blocker (such as amlodipine) may be used pre-operatively in some people.[107] This will not drastically lower the patients blood pressure and make them hypotensive, but it will assist the surgical and anesthesia teams if there is hemodynamic instability during the operation.
Beta blockade
An elevated heart rate (tachycardia) and the feeling of a racing heart (palpitations) may follow after initiating an alpha-adrenoceptor antagonist. If that is the case, a beta-adrenoceptor antagonist is then prescribed to control the heart rate.[103] Just as with the alpha antagonists, there are selective (beta-1) and non-selective (beta-1 and beta-2) adrenoceptor antagonists. The selective agents (atenolol, metoprolol) are preferred to the non-selective agents (propranolol).[103] There are several (labetalol, carvedilol) combined alpha-beta-adrenoceptor antagonists. These agents should be avoided whenever possible as there is upwards of seven times more beta-adrenoceptor antagonism than alpha, which can worsen hypertension and lead to a catecholamine crisis.[108]
Complications
Beta-adrenoceptor antagonists should not be given alone in a pheochromocytoma patient - this can lead to severe consequences.[109] In 1995, a team of physicians from London described the death of a person who had been recently diagnosed pheochromocytoma after initiation of propranolol, a non-selective beta blocker. She quickly developed a hypertensive crisis leading to shock, myocardial infarction, heart failure, and dense right hemiplegia. Despite attempts at resuscitation, the person died several days later.[110] This complication is related to the impact that alpha and beta-adrenoceptor antagonists have on blood vessels combined with the actions of catecholamines. The normal blood vessel is open, allowing for adequate blood flow. When catecholamines activate the alpha receptor, the vessel constricts (gets smaller), which results in hypertension.[111] However, when catecholamines active the beta receptor, the blood vessel dilates (gets larger) and allows for increased blood flow, reducing the blood pressure.[112] If a pheochromocytoma patient is only started on a beta-adrenoceptor antagonist, this reverses the protective vasodilation and worsens the patients hypertension.
Controversy
While the pre-operative alpha and beta blockade discussed above is overwhelmingly recognized as the standard of care, particularly in the United States, there has been discussion at the international level if a blockade is necessary. In 2017, a team of researchers from Germany published an observational case series that called into question the current recommendations for a blockade.[113] The study examined the intraoperative maximal systolic arterial pressure in people with and without alpha-adrenoceptor blockade and found no difference in complications between the two groups.[113] The following year, a group from France published a similar article with a warning against waiting an entire week to begin alpha-blockade. The French researchers called for immediate surgical intervention and consideration of steps to mitigate any intraoperative catecholamine crisis.[114] These articles resulted in rebuttals[104][115] from research teams in the United States, but an international consensus has not yet been reached.
Perioperative fluid status
Excess catecholamines cause a decrease in the total blood volume, making a patient vulnerable to hypotension during the operation.[116] Therefore, a high-sodium diet with adequate fluid intake should be encouraged prior to surgery.[117] Some institutions in the United States will even admit patients the night prior to surgery for intravenous fluid replacement starting at midnight until the time of the operation.[103] However, a small trial from 2009 reported no difference in mortality in patients treated with preoperative intravenous fluids compared to those who did not.[118]
In a 2010 survey of 40 endocrinologists by researchers at the Cedars-Sinai Medical Center in Los Angeles, California, nearly all indicated the importance of preoperative volume resuscitation (having the patient take in plenty of fluids prior to surgery). However, after reviewing their patient data, over 60% of the same physicians failed to discuss salt-loading and adequate hydration.[119] When the patients were stratified by age, those that were younger received the advice to hydrate, but older patients did not. It was hypothesized that the providers chose to forego volume repletion in the older patient population for fear of their potential comorbidities (heart failure) where excess fluid is dangerous.[119] While there is still no recognized consensus or gold standard, providers should individualize the decision based on the patient's perceived nutritional standing, volume status, comorbidities, and ability to self-hydrate.
Post-operative management
The most common post-operative complications, likely causes, and treatment options are:[120][121]
- Hypertension: In the pheochromocytoma patient, postoperative hypertension could indicate incomplete tumor resection or another tumor of unknown location. However, the traditional, non-specific causes of postoperative hypertension including pain, fluid overload, and essential hypertension must also be considered. A perioperative hypertensive crisis is first treated with a 5.0 milligram (mg) intravenous bolus of phentolamine, with additional 5.0 mg dose every ten minutes until the blood pressure falls within an acceptable range.[122] If the blood pressure is only minimally elevated, the patient can resume their alpha and beta-adrenoceptor antagonist from prior to surgery.[120]
- Hypotension: There are several reasons a patient may have low blood pressure in the post-operative period. First and foremost, the tumor (and its abundance of catecholamines causing high blood pressure) has been removed. Furthermore, the patient may still experience the effects of their alpha-adrenoceptor antagonist, which causes lower blood pressure.[121] First-line treatment for postoperative hypotension is aggressive fluid resuscitation, which is why ensuring the patient is well-hydrated (see above) prior to surgery is so imperative.[120] Vasopressors may be needed if the blood pressure does not respond to fluids.
- Hyperglycemia: Catecholamines prevent the secretion of insulin – a hormone responsible for lowering the body's blood glucose (sugar). Blood glucose levels should be checked frequently in the perioperative period and insulin should be given as needed if levels are elevated. Following resection, tumor-related hyperglycemia is likely to resolve.
- Hypoglycemia: After the tumor is removed, insulin is no longer inhibited, which can bring the blood glucose dangerously low. Symptoms include tremor, anxiety, palpitations, sweating, altered mental status (confusion), dizziness, and blurred vision.[123] A retrospective analysis of beta blocker found that some beta blocker use may cause people to more prone to hypoglycemia and not experience these symptoms, which could delay the diagnosis.[124]
- Adrenal Insufficiency: Following a bilateral adrenalectomy (left and right), the patient is no longer capable of secreting the necessary hormones to keep their body functioning. Life-long steroid (hydrocortisone and fludrocortisone) oral supplementation may be required to ensure they do not develop adrenal insufficiency.[125] When the body is stressed (during surgery), the adrenal glands naturally produce more steroids; however, if the glands have been removed, they are unable to do so. Therefore, "stress-dosing" steroids are required and should be started intraopertively to mimic the natural physiology of the adrenal glands.[126] The typical regimen when post-operative adrenal insufficiency is thought to be likely:[120][121]
- 50 milligram (mg) intravenous hydrocortisone in the operating room prior to anesthesia
- Repeat administration of 25–50 mg intravenous hydrocortisone every eight hours for a maximum of 72 hours (3 days) after the operation. Convert to oral replacement therapy as soon as the patient is able to take medication by mouth
- Patients should be transitioned to a normal maintenance (regular, daily) dose of steroids prior to discharge and referred to endocrinology for proper titration and management. Depending on the patient's total body surface area, the total typical daily dose of hydrocortisone is between 15 and 25 mg daily (divided into morning and afternoon pills).[127]
- Those who have lost both their adrenal glands will also require another steroid (mineralcorticoid replacement). The typical daily dose is between 50 and 200 micrograms of fludrocortisone[127]
There have been many other reported complications (renal failure, heart failure, intestinal pseudo-obstruction) following tumor resection. However, the above are more likely to be encountered, which is why their management has been specifically outlined here in this article.
Metastatic disease
Diagnosis and location
Metastatic pheochromocytoma is defined as the presence of tumor cells (chromaffin tissue) where they are not normally found.[128] Patients with a paraganglioma are more likely to develop metastases than those with a pheochromocytoma.[129] The most common extra-adrenal sites of metastases are the lymph nodes, lung, liver, and bone.[130] There have been several studied risk factors associated with the development of metastatic disease - while the patients genetic background plays an important role, the initial age of presentation and size of the tumor lead to negative outcomes.[128] Of all the genetic variants, succinate dehydrogenase subunit B (SDHB) mutations have the highest rates of developing metastatic disease.[129] Another study has reported increased mortality associated with male sex and synchronous metastases.[129] Metastases are divided into synchronous and metachronous; those that are synchronous have developed within several months of the primary tumor, while metachronous metastases do not appear for a significant period of time.[131]
Laparoscopic approach to the original disease, especially in big tumors, has been appointed as an important risk factor for tumoral seeding.[132]
Despite all of the below potential treatment options, recent literature highlights that (for most patients) metastatic pheochromocytoma is slow-growing. In patients with minimal disease burden, a "watch and wait" approach with frequent imaging to monitor disease is favorable, withholding treatment until evidence of progression is visualized.[133]
Treatment
Metastatic pheochromocytoma is best managed with a multidisciplinary team of oncologists, surgeons, radiologists, nuclear medicine physicians, and endocrinologists. There are several treatment options available to patients depending on the amount and location of disease:
Surgery - Normally, the goal of surgery is complete cytoreductive surgery;[132] leave no remnant of disease.[134] However, with widespread metastatic disease, this is not always feasible. Therefore, a surgical debulking procedure is performed (removing as much of the cancerous tissue as possible) in order to reduce patient symptoms by removing the source of catecholamines, improve response to chemo or radionuclide therapy, or simply decrease the size of the tumor.[135] Unfortunately, the intended relief from the procedure is often short-lived, especially if the patient has disease outside the abdomen.[135] A 2013 study from the National Institutes of Health reported that a majority of patients with recurrent biochemical evidence of disease within one year of the operation and less than 30% continued to be biochemically free of disease after five years.[135]
In contrast to an operation for non-metastatic disease, an open procedure may be preferred over a minimally invasive technique in order to circumvent potential tumor spread.[136] This also aids surgical visualization and offers the best opportunity to identify and remove metastatic lymph nodes.[137] Reports have also indicated the utility of administering a radionuclide agent like iodine-123 meta-iodobenzylguanadine (123I-MIBG) prior to surgery and then scanning the patient intraoperatively with a probe to detect disease that may be missed with the naked eye.[138]
Radiation Therapy - With regard to pheochromocytoma, radiation techniques are primarily used for pain control, specifically with regards to bone metastases, local control of the disease, and to limit spinal cord compression.[139] A multidisciplinary team from the Mayo Clinic retrospectively reviewed all of their patients who underwent external beam radiation therapy from 1973 to 2015 and reported that 94% of patients acknowledged symptomatic improvement and over 80% of patients showed no evidence of recurrent disease 5-years post-therapy.[140] Another report from the same institution looked at almost two decades of patients who underwent radiofrequency ablation, cryoablation, or percutaneous ethanol injection for metastatic pheochromocytoma and reported that local control was achieved in over 85% of targeted lesions and that 92% of procedures were associated with reduced pain and/or symptoms of catecholamine excess.[141]
Chemotherapy - The most common chemotherapy regimen for metastatic pheochromocytoma is cyclophosphamide, vincristine, and dacarbazine, collectively known as CVD.[142][143] Response to therapy is measured by a reduction in total tumor volume as well as symptomatic relief, reported by the patient. A systematic review and meta-analysis of unstratified pheochromocytoma patients who underwent CVD therapy showed that 37% of patients had a significant reduction in tumor volume, while 40% of patients experienced lower catecholamine burden.[142] While there was no difference in overall survival between patients whose tumors shrunk versus those without a response (no reduction in tumor burden via imaging), even in non-responders, patients reported feeling better, blood pressure was lower, and some patients were even able to undergo surgery following disease stabilization with CVD.[144] When patients are studied by various categories, research has suggested that females are less likely to have extended survival with CVD chemotherapy compared to their male counterparts.[145] Genetic status has been shown to greatly impact response to CVD. A team of researchers from the National Institutes of Health reported that patient's with succinate dehydrogenase subunit B (SDHB) mutations are not only more likely to initially respond to CVD, but that they also experienced over 30 months of progression free survival (time until tumor returned) with continued administration.[146]
However, CVD is not the only proven chemotherapeutic regimen in the pheochromocytoma patient. A 2018 report demonstrated the remarkable response of two SDHB patients who failed CVD chemotherapy (disease progressed despite medication), but were then treated with temozolomide (TMZ) and had progression free survival of 13 and 27 months, indicating that TMZ can be considered as an alternative treatment regimen in those who have progressed on CVD.[147] Several studies have since reported successful responses with TMZ, particularly in the SDHB sub-population.[148][149]
Radionuclide Therapy
- Iodine-131 meta-iodobenzylguanadine (MIBG)
- As was mentioned in the functional imaging section above, MIBG is not only useful in locating the presence of metastatic disease, but also as an available treatment modality. In 2019, a multi-center phase 2 trial looked at the safety and efficacy of MIBG therapy in metastatic or unresectable (not conducive to surgery) pheochromocytoma patients and the results were promising.[150] Median overall survival was 36.7 months and 92% of patients had at least a partial positive response (tumor shrinkage) or stable disease without progression within the first year of the study. Furthermore, over a fourth of the patients were able to decrease their anti-hypertensive medications and reported symptomatic improvement.[150] There are several patients who are not eligible for MIBG treatment, including pregnant women (exposure to radiation is harmful to the fetus), women who are actively breast feeding, patients in renal failure, and those are who not expected to live longer than 3-months.[151] As MIBG therapy can destroy the thyroid, protective medications (potassium iodide) are started prior to treatment and need to be continued for at least 3 weeks after therapy concludes.[151] Associated side-effects (muscle weakness, nausea, vomiting and hematologic (blood) toxicities, are common, but often minimal, and can be mitigated with slow, steady dosing.[152]
- Peptide Receptor Radionuclide Therapy (PRRT)
- The newest of the treatment options, PRRT utilizes the 68-Ga DOTA analogues mentioned above in the functional imaging section.[153] Treatment with 177Lu-DOTATATE first demonstrated success in patients with undifferentiated neuroendocrine tumors and then trials began with metastatic pheochromocytoma patients.[154][155] In 2019, Vyakaranam et al. published favourable results for their 22 patients who underwent PRRT, with partial response in 2 patients and stable disease (no progression) in the remaining 20 patients.[156] Overall toxicity was low, with no high-grade haematological (blood) or kidney damage reported.[156] At the end of that same year, a systemic review looked at all published articles (12) where metastatic pheochromocytoma patients underwent PRRT and found that treatment-related adverse events are minimal, with only 5 out of 102 patients choosing to voluntarily initiate treatment discontinuation.[157] Newer reports have detailed the utility of combining 90Y-DOTATATE with the traditionally studied 177Lu analog and the various possibilities and novel treatment options these combinations will bring to the field.[95] While the overall reported side-effects have been promising, it is important While the overall reported side-effects have been promising, it is important to note that a collaborative effort between the National Institutes of Health and Radboud University Medical Centre reported two unfortunate cases of rapid disease progression following a remarkable, almost complete response to PRRT. While the etiology of their recurrence is unknown, the team speculated that an elevated tumor marker (Ki-67) could be an indication of a poor response to PRRT and called for pre-PRRT assessments to include Ki-67 values to help individualize patient treatment plans.[158]
Prognosis
According to the National Cancer Institute, prognosis is defined as the likely outcome of a disease OR, the chance of recovery or a recurrence.[159] This is an extremely difficult question when it comes to pheochromcytoma, and the answer depends on the patients genetic status, presence of metastatic disease, and the location of their primary tumor.[160] An article about prognosis published in 2000 reported a 91% 5-year survival rate in their patient population; however, it is important to note that over 86% of their patients had sporadic tumors (no known genetic mutation), which commonly have low malignant potential.[161] In 2019, a consortium of almost twenty European medical centers looked at the prognosis of malignant pheochromocytoma and the data starkly varies from the report of sporadic, single tumors, with a median survival of 6.7 years.[162] Overall survival improved if the patient had (1) disease of the head and neck compared to abdomen, (2) less than 40 years of age, (3) and if their biochemistry was less than five times the upper reference limit of normal.[162]
Recent literature has detailed several factors that predict accelerated progression of disease and higher mortality rates, including patients who choose to forego surgical resection of their primary tumor, larger tumors at initial presentation, older age at initial diagnosis, and a shortened time from primary tumor to presence of metastases.[163] The actual location of the metastases can also indicate prognosis, with osseous lesions (bone) faring better than their soft-tissue (lung, liver) counterparts.[164]
Epidemiology
According to the North American Neuroendocrine Tumor Society, the prevalence of pheochromocytoma is between 1:2500 and 1:6500, meaning that for every 2,500 – 6,500 people, there is (on average) one person with pheochromocytoma.[165] In the United States, this equates to an annual incidence (new cases per year) of 500 to 1600 cases.[165] However, approximations in the early 2000s reported that upwards of 50% of pheochromocytoma diagnoses are at autopsy; therefore, the above estimations may be lower than expected.[11] In a 50-year autopsy case series, the Mayo Clinic reviewed 54 pheochromocytoma cases between 1928–1977 and discovered that just 24% of the patients were correctly diagnosed prior to their death. [166] Outside of the United States, several countries have documented their own epidemiological studies and compared them to what is known in North America. In the first national, epidemiological population-based study in Asia utilizing Korean National Health Insurance Service data, the prevalence of a pheochromocytoma was reported at 2.13 per 100,000 persons with an incidence of 0.18 per 100,000 person-years.[167] This is lower than the occurrence reported from Rochester, Minnesota (0.8 per 100,000 person-years) in a study conducted from 1950 to 1979.[2] However, the Netherlands also conducted a study using a nationwide registry and reported incidence results of 0.57 per 100,000 person-years from 2011 to 2015, which was a significant increase from their 0.37 cases per 100,000 person-years reported from 1995 to 1999.[168] Current hypotheses for why the incidence of pheochromocytoma is growing in the Dutch population point to the advent of modern imaging evaluation and the ability to detect these tumors prior to death.[169] While each of the above studies reported varying incidence and prevalence values, all have indicated that the average age at initial diagnosis is between the third to fifth decade of life.[170] When younger patients are diagnosed with a pheochromocytoma, there should be a high suspicion for hereditary disease, as genetic anticipation (earlier disease onset with each generation) is associated with some mutations.[171]

Classically, the pheochromocytoma "rules of 10" have been taught, particularly to medical students:[172]
- 10% of patients have malignant disease
- 10% of patients have bilateral (both left and right adrenal glands) disease
- 10% of patients have extra-adrenal (paraganglioma) disease
- 10% of patients have inherited (familial disease)
Despite the prominence in many respected textbooks, these guidelines have since been established as inaccurate and are not used in current epidemiological discussions.[170]
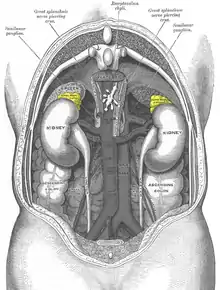
As suggested above, incidental imaging has become a major player in the diagnosis of patients with pheochromocytoma, with current estimates between 10 and 49% of all cases diagnosed after imaging was obtained for another reason. When an adrenal nodule (potential tumor) is discovered on computed tomography or magnetic resonance imaging, there is between a 5 and 10% chance the lesion is a pheochromocytoma.[170] The incidence of adrenal tumors is found in the infographic above, with pheochromocytoma noted in yellow in the top right corner.
History
.jpg.webp)
In 1800, an Irish physician (Charles Sugrue) penned a case report to the London Medical and Physical Journal describing the peculiar case of an 8-year-old male patient who had had seemingly random fits of pain concentrated in the abdomen accompanied by "a hectic flush distinctly marked on each cheek" with a "constant profuse and universal perspiration."[173] Following his death, a group of physicians performed an autopsy to determine cause of death and discovered a six-inch oblong tumor composed of an unknown "yellow-ish coloured substance" coming from the capsula renalis (what is now known as the adrenal gland).[173] This would become the first known clinical description of a pheochromocytoma, but as no features of the tumor itself were described, complete credit is given to the German Felix Fraenkel, who provided a clinical and morphologic picture of this tumor.[174][175] While various physicians were recognizing symptoms and treating patients, Czech biologist Alfred Kohn reported his discovery of the paraganglia system, which would later become crucial to the diagnosis of these tumors. Furthermore, he also introduced the term "chromaffin," allowing pathologists to recognize tumors that arose from the adrenal gland.[176]
In 1908, two pathologists, Henri Alezais and Felix Peyron, introduced the scientific community to "paraganglioma" after they discovered extra-adrenal tissue that reacted to chromium salts, which mimicked the reaction of the adrenal medulla.[177] Just four years later, German pathologist Ludwig Pick coined the term "pheochromocytoma" after he observed the consistent color change in tumors associated with the adrenal medulla.[178] Many surgeons attempted to remove these tumors over the next decade, but their patients died intraoperatively from shock. In 1926, Charles Mayo (a founder of the Mayo Clinic) became the first physician to successfully excise a pheochromocytoma.[178] However, Mayo was likely unaware of the diagnosis prior to the operation. Not until 1929 was a pheochromocytoma recognized preoperatively.[11] Throughout the early 1900s, the operative mortality rate for a pheochromocytoma ranged from 30 to 45%. Retrospective series have postulated that these alarmingly high death rates were due to the lack of a pre-operative blockade with alpha and beta-adrenoceptor antagonist and the need for modern anesthesia practices.[179] From this point forward, physician-scientists have been recognizing patterns in patients with pheochromocytoma and identifying genetic associations and various syndromes.[11]
Society and culture
While a rare disease, there have been several references to pheochromocytoma in popular culture and the media, specifically medical television dramas. Additionally, there is a strong online patient advocacy community that works to connect patients with rare diseases and allows them to meet other individuals who are experiencing similar diagnoses and treatment strategies.
Zebra culture

In the medical community, students are often taught "when you hear hoofbeats, think horses, not zebras."[180] In other words, common diagnoses are common, so healthcare professionals should first rule out what is most expected (the horses) before diving into the rare etiologies that are far less likely to be correct (the zebras). However, the symbol of the zebra has become increasingly powerful to the rare disease community and resulted in several organizations, societies, and special events (Rare Disease Day) to draw attention to the least common option sometimes being the correct diagnosis.[181]
The National Organization for Rare Disorders is a United States-based advocacy parent organization with the goal of promoting awareness and research opportunities to cure rare diseases.[182] Groups such as these encourage patients to become their own advocates and change agents in their healthcare decision-making processes.
Media
In July 2012, an actual pheochromocytoma patient, Tannis Brown, former vice-president of the PheoPara Troopers, was featured on the Discovery Fit & Health Network program Diagnosis: Dead or Alive.[183] The show highlighted her personal struggle with misdiagnosed disease as many physicians felt her episodic headaches and hypertension (high blood pressure) were related to stress.[184]
In the seventh and eighth seasons of Greys Anatomy, series regular Henry has a Von Hippel-Lindau (VHL) mutation that has resulted in a pheochromocytoma. The story arc was met with mixed opinions from the rare disease community.[185] Then executive Director of the VHL Alliance was happy with the portrayal of a VHL patient in mainstream media, but pointed out that of the four scripts she knew of with a VHL patient, three involved a pheochromocytoma, which actually occurs in less than a fifth of all VHL patients.[186][187]
A case of pheochromocytoma was featured in the first episode of season 2 of House MD. Dr. House and his team are tasked with diagnosing and treating an inmate on death row. Although the patient has a violent history of homicide, Dr. House suspects that his episodic rage and aggression may be caused by an adrenaline secreting tumor. Dr. House is able to locate the tumor and diagnoses the patient with pheochromocytoma. Dr. Foreman, one of the doctors, attempts to appeal the inmate's death penalty on the basis that he was unable to control his actions due to his tumor. This kind of legal defense is rarely successful, however.
References
- "PHAEOCHROMOCYTOMA | Meaning & Definition for UK English". Lexico.com. Archived from the original on February 25, 2021. Retrieved 2022-08-24.
- Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT (December 1983). "Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979". Mayo Clinic Proceedings. 58 (12): 802–4. PMID 6645626.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K (20–26 August 2005). "Phaeochromocytoma". Lancet. 366 (9486): 665–75. doi:10.1016/S0140-6736(05)67139-5. PMID 16112304. S2CID 208788653.
- Oyasu R, Yang XJ, Yoshida O, eds. (2008). "What is the difference between pheochromocytoma and paraganglioma? What are the familial syndromes that have pheochromocytoma as a component? What are the pathologic features of pheochromocytoma indicating malignancy?". Questions in Daily Urologic Practice. Questions in Daily Urologic Practice: Updates for Urologists and Diagnostic Pathologists. Tokyo: Springer Japan. pp. 280–284. doi:10.1007/978-4-431-72819-1_49. ISBN 978-4-431-72819-1.
- Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, et al. (March 2002). "Biochemical diagnosis of pheochromocytoma: which test is best?". JAMA. 287 (11): 1427–34. doi:10.1001/jama.287.11.1427. PMID 11903030.
- Moore MG, Netterville JL, Mendenhall WM, Isaacson B, Nussenbaum B (April 2016). "Head and Neck Paragangliomas: An Update on Evaluation and Management". Otolaryngology–Head and Neck Surgery. 154 (4): 597–605. doi:10.1177/0194599815627667. PMID 26861230. S2CID 23547346.
- Williams MD (September 2017). "Paragangliomas of the Head and Neck: An Overview from Diagnosis to Genetics". Head and Neck Pathology. 11 (3): 278–287. doi:10.1007/s12105-017-0803-4. PMC 5550402. PMID 28321772.
- Kellerman RD, Rakel D (2020). Conn's Current Therapy. Elsevier–Health Science. ISBN 978-0-323-79006-2. OCLC 1145315791.
- Tevosian SG, Ghayee HK (December 2019). "Pheochromocytomas and Paragangliomas". Endocrinology and Metabolism Clinics of North America. 48 (4): 727–750. doi:10.1016/j.ecl.2019.08.006. PMID 31655773. S2CID 204947638.
- Zuber SM, Kantorovich V, Pacak K (June 2011). "Hypertension in pheochromocytoma: characteristics and treatment". Endocrinology and Metabolism Clinics of North America. 40 (2): 295–311, vii. doi:10.1016/j.ecl.2011.02.002. PMC 3094542. PMID 21565668.
- Manger WM (August 2006). "An overview of pheochromocytoma: history, current concepts, vagaries, and diagnostic challenges". Annals of the New York Academy of Sciences. 1073 (1): 1–20. Bibcode:2006NYASA1073....1M. doi:10.1196/annals.1353.001. PMID 17102067. S2CID 21423113.
- Hosseinnezhad A, Black RM, Aeddula NR, Adhikari D, Trivedi N (2011). "Glucagon-induced pheochromocytoma crisis". Endocrine Practice. 17 (3): e51-4. doi:10.4158/EP10388.CR. PMID 21324811.
- Lanier JB, Mote MB, Clay EC (September 2011). "Evaluation and management of orthostatic hypotension". American Family Physician. 84 (5): 527–36. PMID 21888303.
- Mitchell L, Bellis F (September 2007). "Phaeochromocytoma--"the great mimic": an unusual presentation". Emergency Medicine Journal. 24 (9): 672–3. doi:10.1136/emj.2007.049569. PMC 2464664. PMID 17711956.
- Harrison's principles of internal medicine. Braunwald, Eugene, 1929– (15th ed.). New York: McGraw-Hill. 2001. ISBN 0-07-913686-9. OCLC 44860874.
{{cite book}}: CS1 maint: others (link) - Zuber SM, Kantorovich V, Pacak K (June 2011). "Hypertension in pheochromocytoma: characteristics and treatment". Endocrinology and Metabolism Clinics of North America. 40 (2): 295–311, vii. doi:10.1016/j.ecl.2011.02.002. PMC 3094542. PMID 21565668.
- Riester A, Weismann D, Quinkler M, Lichtenauer UD, Sommerey S, Halbritter R, et al. (December 2015). "Life-threatening events in patients with pheochromocytoma". European Journal of Endocrinology. 173 (6): 757–64. doi:10.1530/EJE-15-0483. PMID 26346138.
- Prejbisz A, Lenders JW, Eisenhofer G, Januszewicz A (November 2011). "Cardiovascular manifestations of phaeochromocytoma". Journal of Hypertension. 29 (11): 2049–60. doi:10.1097/HJH.0b013e32834a4ce9. PMID 21826022. S2CID 23444609.
- Young WF (December 2007). "Adrenal causes of hypertension: pheochromocytoma and primary aldosteronism". Reviews in Endocrine & Metabolic Disorders. 8 (4): 309–20. doi:10.1007/s11154-007-9055-z. PMID 17914676. S2CID 6009557.
- Liao WB, Liu CF, Chiang CW, Kung CT, Lee CW (September 2000). "Cardiovascular manifestations of pheochromocytoma". The American Journal of Emergency Medicine. 18 (5): 622–5. doi:10.1053/ajem.2000.7341. PMID 10999582.
- Lenders JW (February 2012). "Pheochromocytoma and pregnancy: a deceptive connection". European Journal of Endocrinology. 166 (2): 143–50. doi:10.1530/EJE-11-0528. PMID 21890650.
- Kattah AG, Garovic VD (May 2013). "The management of hypertension in pregnancy". Advances in Chronic Kidney Disease. 20 (3): 229–39. doi:10.1053/j.ackd.2013.01.014. PMC 3925675. PMID 23928387.
- Zhang R, Gupta D, Albert SG (December 2017). "Pheochromocytoma as a reversible cause of cardiomyopathy: Analysis and review of the literature". International Journal of Cardiology. 249: 319–323. doi:10.1016/j.ijcard.2017.07.014. PMID 29121733.
- Agrawal S, Shirani J, Garg L, Singh A, Longo S, Longo A, et al. (March 2017). "Pheochromocytoma and stress cardiomyopathy: Insight into pathogenesis". World Journal of Cardiology. 9 (3): 255–260. doi:10.4330/wjc.v9.i3.255. PMC 5368675. PMID 28400922.
- Van YH, Wang HS, Lai CH, Lin JN, Lo FS (November 2002). "Pheochromocytoma presenting as stroke in two Taiwanese children". Journal of Pediatric Endocrinology & Metabolism. 15 (9): 1563–7. doi:10.1515/jpem.2002.15.9.1563. PMID 12503867. S2CID 37955071.
- Abourazzak S, Atmani S, Arqam LE, Chaouki S, Labib S, Harrandou M, et al. (May 2010). "Cerebral ischaemic stroke and bilateral pheochromocytoma". BMJ Case Reports. 2010: bcr1220092535. doi:10.1136/bcr.12.2009.2535. PMC 3047554. PMID 22736758.
- Dagartzikas MI, Sprague K, Carter G, Tobias JD (February 2002). "Cerebrovascular event, dilated cardiomyopathy, and pheochromocytoma". Pediatric Emergency Care. 18 (1): 33–5. doi:10.1097/00006565-200202000-00011. PMID 11862137. S2CID 44533238.
- Cohen JK, Cisco RM, Scholten A, Mitmaker E, Duh QY (April 2014). "Pheochromocytoma crisis resulting in acute heart failure and cardioembolic stroke in a 37-year-old man". Surgery. 155 (4): 726–7. doi:10.1016/j.surg.2012.11.013. PMID 23305592.
- Lin PC, Hsu JT, Chung CM, Chang ST (2007). "Pheochromocytoma Underlying Hypertension, Stroke, and Dilated Cardiomyopathy". Texas Heart Institute Journal. 34 (2): 244–6. OCLC 679006463. PMC 1894695. PMID 17622380.
- Buchbinder NA, Yu R, Rosenbloom BE, Sherman CT, Silberman AW (December 2009). "Left ventricular thrombus and embolic stroke caused by a functional paraganglioma". Journal of Clinical Hypertension. 11 (12): 734–7. doi:10.1111/j.1751-7176.2009.00182.x. PMC 8673247. PMID 20021531. S2CID 30275458.
- Luiz HV, da Silva TN, Pereira BD, Santos JG, Gonçalves D, Manita I, Portugal J (December 2013). "Malignant paraganglioma presenting with hemorrhagic stroke in a child". Pediatrics. 132 (6): e1709-14. doi:10.1542/peds.2013-0492. PMID 24276837. S2CID 7618637.
- Potapova G, Chazova I, Kuznetsov N, Sitina V, Popov E, Gavrilov I (June 2011). "Pheochromocytoma and Stroke". Journal of Hypertension. 29: e505. doi:10.1097/00004872-201106001-01534.
- Anderson NE, Chung K, Willoughby E, Croxson MS (April 2013). "Neurological manifestations of phaeochromocytomas and secretory paragangliomas: a reappraisal". Journal of Neurology, Neurosurgery, and Psychiatry. 84 (4): 452–7. doi:10.1136/jnnp-2012-303028. PMID 23204473. S2CID 207005321.
- Shemin D, Cohn PS, Zipin SB (November 1990). "Pheochromocytoma presenting as rhabdomyolysis and acute myoglobinuric renal failure". Archives of Internal Medicine. 150 (11): 2384–5. doi:10.1001/archinte.1990.00390220118024. PMID 2241450.
- Hamada N, Akamatsu A, Joh T (January 1993). "A case of pheochromocytoma complicated with acute renal failure and cardiomyopathy". Japanese Circulation Journal. 57 (1): 84–90. doi:10.1253/jcj.57.84. PMID 8437346.
- Celik H, Celik O, Guldiken S, Inal V, Puyan FO, Tugrul A (February 2014). "Pheochromocytoma presenting with rhabdomyolysis and acute renal failure: a case report". Renal Failure. 36 (1): 104–7. doi:10.3109/0886022X.2013.832856. PMID 24059440. S2CID 2062065.
- Takabatake T, Kawabata M, Ohta H, Yamamoto Y, Ishida Y, Hara H, Hattori N (July 1985). "Acute renal failure and transient, massive proteinuria in a case of pheochromocytoma". Clinical Nephrology. 24 (1): 47–9. PMID 4017298.
- Lorz W, Cottier C, Imhof E, Gyr N (1993). "Multiple organ failure and coma as initial presentation of pheochromocytoma in a patient with multiple endocrine neoplasia (MEN) type II A". Intensive Care Medicine. 19 (4): 235–8. doi:10.1007/BF01694777. PMC 7095150. PMID 8103532.
- Marshall JC (2001). "The multiple organ dysfunction syndrome". In Holzheimer RG, Mannick JA (eds.). Surgical Treatment: Evidence-Based and Problem-Oriented. Munich: Zuckschwerdt. ISBN 978-3-88603-714-8.
- Newell KA, Prinz RA, Pickleman J, Braithwaite S, Brooks M, Karson TH, Glisson S (August 1988). "Pheochromocytoma multisystem crisis. A surgical emergency". Archives of Surgery. 123 (8): 956–9. doi:10.1001/archsurg.1988.01400320042007. PMID 2899426.
- Fishbein L (February 2016). "Pheochromocytoma and Paraganglioma: Genetics, Diagnosis, and Treatment". Hematology/Oncology Clinics of North America. 30 (1): 135–50. doi:10.1016/j.hoc.2015.09.006. PMID 26614373.
- Mercado-Asis LB, Wolf KI, Jochmanova I, Taïeb D (January 2018). "Pheochromocytoma: A Genetic and Diagnostic Update" (PDF). Endocrine Practice. 24 (1): 78–90. doi:10.4158/EP-2017-0057. PMID 29144820. S2CID 45860930.
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. (June 2014). "Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline". The Journal of Clinical Endocrinology and Metabolism. 99 (6): 1915–42. doi:10.1210/jc.2014-1498. PMID 24893135.
- Kavinga Gunawardane PT, Grossman A (October 2017). "The clinical genetics of phaeochromocytoma and paraganglioma". Archives of Endocrinology and Metabolism. 61 (5): 490–500. doi:10.1590/2359-3997000000299. PMID 29166454.
- Jochmanova I, Wolf KI, King KS, Nambuba J, Wesley R, Martucci V, et al. (August 2017). "SDHB-related pheochromocytoma and paraganglioma penetrance and genotype–phenotype correlations". Journal of Cancer Research and Clinical Oncology. 143 (8): 1421–1435. doi:10.1007/s00432-017-2397-3. PMC 5505780. PMID 28374168.
- Lahlou-Laforêt K, Consoli SM, Jeunemaitre X, Gimenez-Roqueplo AP (May 2012). "Presymptomatic genetic testing in minors at risk of paraganglioma and pheochromocytoma: our experience of oncogenetic multidisciplinary consultation". Hormone and Metabolic Research. 44 (5): 354–8. doi:10.1055/s-0032-1311568. PMID 22517555.
- Neumann HP, Young WF, Krauss T, Bayley JP, Schiavi F, Opocher G, et al. (August 2018). "65 YEARS OF THE DOUBLE HELIX: Genetics informs precision practice in the diagnosis and management of pheochromocytoma". Endocrine-Related Cancer. 25 (8): T201–T219. doi:10.1530/ERC-18-0085. PMID 29794110.
- Favier J, Amar L, Gimenez-Roqueplo AP (February 2015). "Paraganglioma and phaeochromocytoma: from genetics to personalized medicine". Nature Reviews. Endocrinology. 11 (2): 101–11. doi:10.1038/nrendo.2014.188. PMID 25385035. S2CID 26205361.
- Dahia PL (February 2014). "Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity". Nature Reviews. Cancer. 14 (2): 108–19. doi:10.1038/nrc3648. PMID 24442145. S2CID 31457232.
- Jochmanova I, Pacak K (January 2018). "Genomic Landscape of Pheochromocytoma and Paraganglioma". Trends in Cancer. 4 (1): 6–9. doi:10.1016/j.trecan.2017.11.001. PMC 5819363. PMID 29413423.
- Taïeb D, Yang C, Delenne B, Zhuang Z, Barlier A, Sebag F, Pacak K (May 2013). "First report of bilateral pheochromocytoma in the clinical spectrum of HIF2A-related polycythemia-paraganglioma syndrome". The Journal of Clinical Endocrinology and Metabolism. 98 (5): E908-13. doi:10.1210/jc.2013-1217. PMC 3644612. PMID 23539726.
- Yang C, Sun MG, Matro J, Huynh TT, Rahimpour S, Prchal JT, et al. (March 2013). "Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas". Blood. 121 (13): 2563–6. doi:10.1182/blood-2012-10-460972. PMC 3612863. PMID 23361906.
- Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, et al. (May 2013). "New syndrome of paraganglioma and somatostatinoma associated with polycythemia". Journal of Clinical Oncology. 31 (13): 1690–8. doi:10.1200/JCO.2012.47.1912. PMC 3807138. PMID 23509317.
- Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, et al. (September 2012). "Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia". The New England Journal of Medicine. 367 (10): 922–30. doi:10.1056/NEJMoa1205119. PMC 3432945. PMID 22931260.
- Dmitriev PM, Wang H, Rosenblum JS, Prodanov T, Cui J, Pappo AS, et al. (December 2019). "Vascular Changes in the Retina and Choroid of Patients With EPAS1 Gain-of-Function Mutation Syndrome". JAMA Ophthalmology. 138 (2): 148–155. doi:10.1001/jamaophthalmol.2019.5244. PMC 7042897. PMID 31876943.
- Toledo RA, Qin Y, Cheng ZM, Gao Q, Iwata S, Silva GM, et al. (May 2016). "Recurrent Mutations of Chromatin-Remodeling Genes and Kinase Receptors in Pheochromocytomas and Paragangliomas". Clinical Cancer Research. 22 (9): 2301–10. doi:10.1158/1078-0432.CCR-15-1841. PMC 4854762. PMID 26700204.
- Carney JA (2013). "Carney triad". Frontiers of Hormone Research. 41: 92–110. doi:10.1159/000345672. ISBN 978-3-318-02330-5. PMID 23652673.
- Stratakis CA, Carney JA (July 2009). "The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications". Journal of Internal Medicine. 266 (1): 43–52. doi:10.1111/j.1365-2796.2009.02110.x. PMC 3129547. PMID 19522824.
- Neumann HP, Young WF, Eng C (August 2019). "Pheochromocytoma and Paraganglioma". The New England Journal of Medicine. 381 (6): 552–565. doi:10.1056/NEJMra1806651. PMID 31390501. S2CID 199505276.
- Eisenhofer G, Goldstein DS, Walther MM, Friberg P, Lenders JW, Keiser HR, Pacak K (June 2003). "Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results". The Journal of Clinical Endocrinology and Metabolism. 88 (6): 2656–66. doi:10.1210/jc.2002-030005. PMID 12788870.
- Griffin TP, Casey R, Wall D, Bell M, O'Shea PM (August 2016). "Evaluating the optimum rest period prior to blood collection for fractionated plasma free metanephrines analysis". Practical Laboratory Medicine. 5: 39–46. doi:10.1016/j.plabm.2016.05.001. PMC 5574516. PMID 28856203.
- Lenders JW, Willemsen JJ, Eisenhofer G, Ross HA, Pacak K, Timmers HJ, Sweep CG (February 2007). "Is supine rest necessary before blood sampling for plasma metanephrines?". Clinical Chemistry. 53 (2): 352–4. doi:10.1373/clinchem.2006.076489. PMID 17200132.
- Neary NM, King KS, Pacak K (June 2011). "Drugs and pheochromocytoma—don't be fooled by every elevated metanephrine". The New England Journal of Medicine. 364 (23): 2268–70. doi:10.1056/NEJMc1101502. PMC 4724800. PMID 21651412.
- de Jong WH, Post WJ, Kerstens MN, de Vries EG, Kema IP (June 2010). "Elevated urinary free and deconjugated catecholamines after consumption of a catecholamine-rich diet". The Journal of Clinical Endocrinology and Metabolism. 95 (6): 2851–5. doi:10.1210/jc.2009-2589. PMID 20382681.
- de Jong WH, Eisenhofer G, Post WJ, Muskiet FA, de Vries EG, Kema IP (August 2009). "Dietary influences on plasma and urinary metanephrines: implications for diagnosis of catecholamine-producing tumors". The Journal of Clinical Endocrinology and Metabolism. 94 (8): 2841–9. doi:10.1210/jc.2009-0303. PMID 19567530.
- Alrezk R, Suarez A, Tena I, Pacak K (2018-11-27). "Update of Pheochromocytoma Syndromes: Genetics, Biochemical Evaluation, and Imaging". Frontiers in Endocrinology. 9: 515. doi:10.3389/fendo.2018.00515. PMC 6277481. PMID 30538672.
- Martucci VL, Pacak K (January 2014). "Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment". Current Problems in Cancer. 38 (1): 7–41. doi:10.1016/j.currproblcancer.2014.01.001. PMC 3992879. PMID 24636754.
- d'Herbomez M, Do Cao C, Vezzosi D, Borzon-Chasot F, Baudin E (September 2010). "Chromogranin A assay in clinical practice". Annales d'Endocrinologie. 71 (4): 274–80. doi:10.1016/j.ando.2010.04.004. PMID 20538257.
- Grossrubatscher E, Dalino P, Vignati F, Gambacorta M, Pugliese R, Boniardi M, et al. (September 2006). "The role of chromogranin A in the management of patients with phaeochromocytoma". Clinical Endocrinology. 65 (3): 287–93. doi:10.1111/j.1365-2265.2006.02591.x. PMID 16918946. S2CID 19506144.
- Därr R, Lenders JW, Stange K, Kindel B, Hofbauer LC, Bornstein SR, Eisenhofer G (January 2013). "[Diagnosis of pheochromocytoma and paraganglioma: the clonidine suppression test in patients with borderline elevations of plasma free normetanephrine]". Deutsche Medizinische Wochenschrift. 138 (3): 76–81. doi:10.1055/s-0032-1327395. PMID 23299341.
- Kosman ME (July 1975). "Evaluation of clonidine hydrochloride (Catapres). A new antihypertensive agent". JAMA. 233 (2): 174–6. doi:10.1001/jama.1975.03260020060030. PMID 1173448.
- England JF (May 1977). "Clonidine rebound hypertension". The Medical Journal of Australia. 1 (20): 756–7. doi:10.5694/j.1326-5377.1977.tb131095.x. PMID 875850. S2CID 67982391.
- Geyskes GG, Boer P, Dorhout Mees EJ (January 1979). "Clonidine withdrawal. Mechanism and frequency of rebound hypertension". British Journal of Clinical Pharmacology. 7 (1): 55–62. doi:10.1111/j.1365-2125.1979.tb00897.x. PMC 1429594. PMID 760743.
- Malaty J, Malaty IA (October 2014). "Hypertensive urgency: an important aetiology of rebound hypertension". BMJ Case Reports. 2014: bcr2014206022. doi:10.1136/bcr-2014-206022. PMC 4208112. PMID 25336552.
- Rao D, Peitzsch M, Prejbisz A, Hanus K, Fassnacht M, Beuschlein F, et al. (August 2017). "Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma". European Journal of Endocrinology. 177 (2): 103–113. doi:10.1530/EJE-17-0077. PMC 5488393. PMID 28476870.
- Lenders JW, Eisenhofer G (June 2017). "Update on Modern Management of Pheochromocytoma and Paraganglioma". Endocrinology and Metabolism. 32 (2): 152–161. doi:10.3803/EnM.2017.32.2.152. PMC 5503859. PMID 28685506.
- Gupta G, Pacak K (June 2017). "Precision Medicine: An Update on Genotype/Biochemical Phenotype Relationships in Pheochromocytoma/Paraganglioma Patients". Endocrine Practice. 23 (6): 690–704. doi:10.4158/EP161718.RA. PMC 7470624. PMID 28332883.
- Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, et al. (April 2005). "Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines". Clinical Chemistry. 51 (4): 735–44. doi:10.1373/clinchem.2004.045484. PMID 15718487.
- Histed SN, Lindenberg ML, Mena E, Turkbey B, Choyke PL, Kurdziel KA (April 2012). "Review of functional/anatomical imaging in oncology". Nuclear Medicine Communications. 33 (4): 349–61. doi:10.1097/MNM.0b013e32834ec8a5. PMC 3295905. PMID 22314804.
- Neumann HP, Young WF, Eng C (August 2019). Longo DL (ed.). "Pheochromocytoma and Paraganglioma". The New England Journal of Medicine. 381 (6): 552–565. doi:10.1056/NEJMra1806651. PMID 31390501. S2CID 199505276.
- Timmers HJ, Taieb D, Pacak K (May 2012). "Current and future anatomical and functional imaging approaches to pheochromocytoma and paraganglioma". Hormone and Metabolic Research. 44 (5): 367–72. doi:10.1055/s-0031-1299712. PMC 4714588. PMID 22399235.
- McCullough PA, Choi JP, Feghali GA, Schussler JM, Stoler RM, Vallabahn RC, Mehta A (September 2016). "Contrast-Induced Acute Kidney Injury". Journal of the American College of Cardiology. 68 (13): 1465–1473. doi:10.1016/j.jacc.2016.05.099. PMID 27659469.
- Caraiani C, Dong Y, Rudd AG, Dietrich CF (December 2018). "Reasons for inadequate or incomplete imaging techniques". Medical Ultrasonography. 20 (4): 498–507. doi:10.11152/mu-1736. PMID 30534659.
- Castinetti F, Kroiss A, Kumar R, Pacak K, Taieb D (August 2015). "15 YEARS OF PARAGANGLIOMA: Imaging and imaging-based treatment of pheochromocytoma and paraganglioma". Endocrine-Related Cancer. 22 (4): T135-45. doi:10.1530/ERC-15-0175. PMID 26045470.
- Leung K, Stamm M, Raja A, Low G (February 2013). "Pheochromocytoma: the range of appearances on ultrasound, CT, MRI, and functional imaging". AJR. American Journal of Roentgenology. 200 (2): 370–8. doi:10.2214/AJR.12.9126. PMID 23345359.
- Chaudhary V, Bano S (September 2012). "Anatomical and functional imaging in endocrine hypertension". Indian Journal of Endocrinology and Metabolism. 16 (5): 713–21. doi:10.4103/2230-8210.100659. PMC 3475894. PMID 23087854.
- Rufini V, Treglia G, Perotti G, Giordano A (January 2013). "The evolution in the use of MIBG scintigraphy in pheochromocytomas and paragangliomas". Hormones. 12 (1): 58–68. doi:10.1007/bf03401287. PMID 23624132. S2CID 4716903.
- van Hulsteijn LT, Niemeijer ND, Dekkers OM, Corssmit EP (April 2014). "(131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis". Clinical Endocrinology. 80 (4): 487–501. doi:10.1111/cen.12341. PMID 24118038. S2CID 38456445.
- Santhanam P, Taïeb D (December 2014). "Role of (18) F-FDOPA PET/CT imaging in endocrinology". Clinical Endocrinology. 81 (6): 789–98. doi:10.1111/cen.12566. PMID 25056984. S2CID 204992362.
- Taïeb D, Tessonnier L, Sebag F, Niccoli-Sire P, Morange I, Colavolpe C, et al. (2008). "The role of 18F-FDOPA and 18F-FDG-PET in the management of malignant and multifocal phaeochromocytomas". Clinical Endocrinology. 69 (4): 580–586. doi:10.1111/j.1365-2265.2008.03257.x. OCLC 798350389. PMID 18394015. S2CID 205284382.
- Taïeb D, Hicks RJ, Hindié E, Guillet BA, Avram A, Ghedini P, et al. (September 2019). "European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma". European Journal of Nuclear Medicine and Molecular Imaging. 46 (10): 2112–2137. doi:10.1007/s00259-019-04398-1. PMC 7446938. PMID 31254038. S2CID 195738862.
- Jha A, Ling A, Millo C, Gupta G, Viana B, Lin FI, et al. (May 2018). "18F-FDG and anatomic imaging in the detection of succinate dehydrogenase mutation (SDHx )-related pheochromocytoma and paraganglioma in the pediatric population". European Journal of Nuclear Medicine and Molecular Imaging. 45 (5): 787–797. doi:10.1007/s00259-017-3896-9. PMC 6707509. PMID 29204718.
- Janssen I, Chen CC, Millo CM, Ling A, Taieb D, Lin FI, et al. (September 2016). "PET/CT comparing (68)Ga-DOTATATE and other radiopharmaceuticals and in comparison with CT/MRI for the localization of sporadic metastatic pheochromocytoma and paraganglioma". European Journal of Nuclear Medicine and Molecular Imaging. 43 (10): 1784–91. doi:10.1007/s00259-016-3357-x. PMC 8194362. PMID 26996779. S2CID 23005709.
- Kroiss AS, Uprimny C, Shulkin BL, Gruber L, Frech A, Jazbec T, et al. (March 2019). "18F-DOPA PET/CT". Revista Espanola de Medicina Nuclear e Imagen Molecular. 38 (2): 94–99. doi:10.1016/j.remn.2018.09.004. PMID 30630744. S2CID 196533632.
- Mak IY, Hayes AR, Khoo B, Grossman A (2019). "Peptide Receptor Radionuclide Therapy as a Novel Treatment for Metastatic and Invasive Phaeochromocytoma and Paraganglioma". Neuroendocrinology. 109 (4): 287–298. doi:10.1159/000499497. PMID 30856620. S2CID 75140335.
- Vyakaranam, Achyut Ram; Crona, Joakim; Norlén, Olov; Hellman, Per; Sundin, Anders (June 2019). "11C-hydroxy-ephedrine-PET/CT in the Diagnosis of Pheochromocytoma and Paraganglioma". Cancers. 11 (6): 847. doi:10.3390/cancers11060847. PMC 6627429. PMID 31248124.
- Wiseman D, Lakis ME, Nilubol N (July 2019). "Precision Surgery for Pheochromocytomas and Paragangliomas". Hormone and Metabolic Research. 51 (7): 470–482. doi:10.1055/a-0926-3618. PMC 8572371. PMID 31307109.
- Aggeli C, Nixon AM, Parianos C, Vletsis G, Papanastasiou L, Markou A, et al. (October 2017). "Surgery for pheochromocytoma: A 20-year experience of a single institution". Hormones. 16 (4): 388–395. doi:10.14310/horm.2002.1759. PMID 29518759. S2CID 4730354.
- Goffredo P, Adam MA, Thomas SM, Scheri RP, Sosa JA, Roman SA (August 2015). "Patterns of Use and Short-Term Outcomes of Minimally Invasive Surgery for Malignant Pheochromocytoma: A Population-Level Study". World Journal of Surgery. 39 (8): 1966–73. doi:10.1007/s00268-015-3040-6. PMID 25821949. S2CID 9017845.
- Berber E, Mitchell J, Milas M, Siperstein A (August 2010). "Robotic posterior retroperitoneal adrenalectomy: operative technique". Archives of Surgery. 145 (8): 781–4. doi:10.1001/archsurg.2010.148. PMID 20713932.
- Neumann HP, Tsoy U, Bancos I, Amodru V, Walz MK, Tirosh A, et al. (August 2019). "Comparison of Pheochromocytoma-Specific Morbidity and Mortality Among Adults With Bilateral Pheochromocytomas Undergoing Total Adrenalectomy vs Cortical-Sparing Adrenalectomy". JAMA Network Open. 2 (8): e198898. doi:10.1001/jamanetworkopen.2019.8898. PMC 6692838. PMID 31397861.
- Lee JE, Curley SA, Gagel RF, Evans DB, Hickey RC (December 1996). "Cortical-sparing adrenalectomy for patients with bilateral pheochromocytoma". Surgery. 120 (6): 1064–70, discussion 1070–1. doi:10.1016/S0039-6060(96)80056-0. PMID 8957496.
- Pacak K (November 2007). "Preoperative management of the pheochromocytoma patient". The Journal of Clinical Endocrinology and Metabolism. 92 (11): 4069–79. doi:10.1210/jc.2007-1720. PMID 17989126.
- Wolf KI, Santos JR, Pacak K (January 2019). "Why Take the Risk? We Only Live Once: The Dangers Associated with Neglecting a Pre-Operative Alpha Adrenoceptor Blockade in Pheochromocytoma Patients". Endocrine Practice. 25 (1): 106–108. doi:10.4158/EP-2018-0455. PMC 6478021. PMID 30289301.
- Graham RM, Thornell IR, Gain JM, Bagnoli C, Oates HF, Stokes GS (November 1976). "Prazosin: the first-dose phenomenon". British Medical Journal. 2 (6047): 1293–4. doi:10.1136/bmj.2.6047.1293. PMC 1689975. PMID 793676.
- Kleeman FJ (June 1977). "Phenoxybenzamine". The Journal of Urology. 117 (6): 814. doi:10.1016/s0022-5347(17)58643-7. PMID 875171.
- Brunaud L, Boutami M, Nguyen-Thi PL, Finnerty B, Germain A, Weryha G, et al. (December 2014). "Both preoperative alpha and calcium channel blockade impact intraoperative hemodynamic stability similarly in the management of pheochromocytoma". Surgery. 156 (6): 1410–7, discussion1417-8. doi:10.1016/j.surg.2014.08.022. PMID 25456922.
- Clark BK (May 1992). "Beta-adrenergic blocking agents: their current status". AACN Clinical Issues in Critical Care Nursing. 3 (2): 447–60. doi:10.4037/15597768-1992-2016. PMID 1349490.
- Luiz HV, Tanchee MJ, Pavlatou MG, Yu R, Nambuba J, Wolf K, et al. (July 2016). "Are patients with hormonally functional phaeochromocytoma and paraganglioma initially receiving a proper adrenoceptor blockade? A retrospective cohort study". Clinical Endocrinology. 85 (1): 62–9. doi:10.1111/cen.13066. PMC 4899243. PMID 26998836.
- Sheaves R, Chew SL, Grossman AB (January 1995). "The dangers of unopposed beta-adrenergic blockade in phaeochromocytoma". Postgraduate Medical Journal. 71 (831): 58–9. doi:10.1136/pgmj.71.831.58-a. PMC 2397901. PMID 7708599.
- van Brummelen P, Jie K, van Zwieten PA (1986). "Alpha-adrenergic receptors in human blood vessels". British Journal of Clinical Pharmacology. 21 Suppl 1 (Suppl 1): 33S–39S. doi:10.1111/j.1365-2125.1986.tb02851.x. PMC 1400759. PMID 2871855.
- Chruscinski A, Brede ME, Meinel L, Lohse MJ, Kobilka BK, Hein L (November 2001). "Differential distribution of beta-adrenergic receptor subtypes in blood vessels of knockout mice lacking beta(1)- or beta(2)-adrenergic receptors". Molecular Pharmacology. 60 (5): 955–62. doi:10.1124/mol.60.5.955. PMID 11641423.
- Groeben H, Nottebaum BJ, Alesina PF, Traut A, Neumann HP, Walz MK (February 2017). "Perioperative α-receptor blockade in phaeochromocytoma surgery: an observational case series". British Journal of Anaesthesia. 118 (2): 182–189. doi:10.1093/bja/aew392. PMID 28100521. S2CID 5979863.
- Lentschener C, Baillard C, Dousset B, Gaujoux S (February 2019). "Dogma is Made to be Broken. Why Are We Postponing Curative Surgery to Administer Ineffective Alpha Adrenoreceptor Blockade in Most Patients Undergoing Pheochromocytoma Removal?". Endocrine Practice. 25 (2): 199. doi:10.4158/1934-2403-25.2.199. PMID 30817194. S2CID 73480157.
- Santos JR, Wolf KI, Pacak K (February 2019). "A Necessity, Not a Second Thought: Pre-Operative Alpha-Adrenoceptor Blockade in Pheochromocytoma Patients". Endocrine Practice. 25 (2): 200–201. doi:10.4158/1934-2403-25.2.200. PMC 7451406. PMID 30817195.
- Challis BG, Casey RT, Simpson HL, Gurnell M (February 2017). "Is there an optimal preoperative management strategy for phaeochromocytoma/paraganglioma?". Clinical Endocrinology. 86 (2): 163–167. doi:10.1111/cen.13252. PMID 27696513. S2CID 1473367.
- Jiang M, Ding H, Liang Y, Tang J, Lin Y, Xiang K, et al. (March 2018). "Preoperative risk factors for haemodynamic instability during pheochromocytoma surgery in Chinese patients". Clinical Endocrinology. 88 (3): 498–505. doi:10.1111/cen.13544. PMID 29292527. S2CID 46820948.
- Lentschener C, Gaujoux S, Thillois JM, Duboc D, Bertherat J, Ozier Y, Dousset B (April 2009). "Increased arterial pressure is not predictive of haemodynamic instability in patients undergoing adrenalectomy for phaeochromocytoma". Acta Anaesthesiologica Scandinavica. 53 (4): 522–7. doi:10.1111/j.1399-6576.2008.01894.x. PMID 19239408. S2CID 25480499.
- Wong C, Yu R (July 2010). "Preoperative preparation for pheochromocytoma resection: physician survey and clinical practice". Experimental and Clinical Endocrinology & Diabetes. 118 (7): 400–4. doi:10.1055/s-0029-1225339. PMID 19609840.
- Mamilla D, Araque KA, Brofferio A, Gonzales MK, Sullivan JN, Nilubol N, Pacak K (July 2019). "Postoperative Management in Patients with Pheochromocytoma and Paraganglioma". Cancers. 11 (7): 936. doi:10.3390/cancers11070936. PMC 6678461. PMID 31277296.
- Naranjo J, Dodd S, Martin YN (August 2017). "Perioperative Management of Pheochromocytoma". Journal of Cardiothoracic and Vascular Anesthesia. 31 (4): 1427–1439. doi:10.1053/j.jvca.2017.02.023. PMID 28392094.
- Aronow WS (May 2017). "Treatment of hypertensive emergencies". Annals of Translational Medicine. 5 (Suppl 1): S5. doi:10.21037/atm.2017.03.34. PMC 5440310. PMID 28567387.
- Iqbal A, Heller S (June 2016). "Managing hypoglycaemia" (PDF). Best Practice & Research. Clinical Endocrinology & Metabolism. 30 (3): 413–30. doi:10.1016/j.beem.2016.06.004. PMID 27432075.
- Dungan K, Merrill J, Long C, Binkley P (November 2019). "Effect of beta blocker use and type on hypoglycemia risk among hospitalized insulin requiring patients". Cardiovascular Diabetology. 18 (1): 163. doi:10.1186/s12933-019-0967-1. PMC 6882013. PMID 31775749.
- Shen WT, Lee J, Kebebew E, Clark OH, Duh QY (August 2006). "Selective use of steroid replacement after adrenalectomy: lessons from 331 consecutive cases". Archives of Surgery. 141 (8): 771–4, discussion 774–6. doi:10.1001/archsurg.141.8.771. PMID 16924084.
- MacKenzie CR, Goodman SM (July 2016). "Stress Dose Steroids: Myths and Perioperative Medicine". Current Rheumatology Reports. 18 (7): 47. doi:10.1007/s11926-016-0595-7. PMID 27351679. S2CID 22000392.
- Pazderska A, Pearce SH (June 2017). "Adrenal insufficiency – recognition and management". Clinical Medicine. 17 (3): 258–262. doi:10.7861/clinmedicine.17-3-258. PMC 6297573. PMID 28572228.
- Zelinka T, Musil Z, Dušková J, Burton D, Merino MJ, Milosevic D, et al. (October 2011). "Metastatic pheochromocytoma: does the size and age matter?". European Journal of Clinical Investigation. 41 (10): 1121–8. doi:10.1111/j.1365-2362.2011.02518.x. PMC 3170415. PMID 21692797.
- Hamidi O, Young WF, Gruber L, Smestad J, Yan Q, Ponce OJ, et al. (November 2017). "Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis". Clinical Endocrinology. 87 (5): 440–450. doi:10.1111/cen.13434. PMC 5854189. PMID 28746746.
- "Metastatic catecholamine-secreting paraganglioma (extra-adrenal pheochromocytoma)". The American Journal of Medicine. 61 (4): 523–532. October 1976. doi:10.1016/0002-9343(76)90332-6. ISSN 0002-9343. PMID 973646.
- Engstrand J, Strömberg C, Nilsson H, Freedman J, Jonas E (December 2019). "Synchronous and metachronous liver metastases in patients with colorectal cancer-towards a clinically relevant definition". World Journal of Surgical Oncology. 17 (1): 228. doi:10.1186/s12957-019-1771-9. PMC 6933908. PMID 31878952.
- Ferrer-Inaebnit, Ester; Segura-Sampedro, Juan José; Alfonso-García, María; González-Argente, Xavier; Morales-Soriano, Rafael (January 2021). "Cytoreductive surgery in functioning peritoneal pheochromocytomatosis". Cirugia Espanola. 99 (1): 73–76. doi:10.1016/j.ciresp.2020.03.010. ISSN 1578-147X. PMID 32402418. S2CID 219405349.
- Corssmit EP, Snel M, Kapiteijn E (January 2020). "Malignant pheochromocytoma and paraganglioma: management options". Current Opinion in Oncology. 32 (1): 20–26. doi:10.1097/cco.0000000000000589. PMID 31599769. S2CID 204029843.
- Roman-Gonzalez A, Zhou S, Ayala-Ramirez M, Shen C, Waguespack SG, Habra MA, et al. (July 2018). "Impact of Surgical Resection of the Primary Tumor on Overall Survival in Patients With Metastatic Pheochromocytoma or Sympathetic Paraganglioma". Annals of Surgery. 268 (1): 172–178. doi:10.1097/sla.0000000000002195. PMID 28257320. S2CID 22915608.
- Ellis RJ, Patel D, Prodanov T, Sadowski S, Nilubol N, Adams K, et al. (September 2013). "Response after surgical resection of metastatic pheochromocytoma and paraganglioma: can postoperative biochemical remission be predicted?". Journal of the American College of Surgeons. 217 (3): 489–96. doi:10.1016/j.jamcollsurg.2013.04.027. PMC 3770940. PMID 23891076.
- Jimenez C, Rohren E, Habra MA, Rich T, Jimenez P, Ayala-Ramirez M, Baudin E (August 2013). "Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma". Current Oncology Reports. 15 (4): 356–71. doi:10.1007/s11912-013-0320-x. PMID 23674235. S2CID 1167562.
- Pappachan JM, Raskauskiene D, Sriraman R, Edavalath M, Hanna FW (July 2014). "Diagnosis and management of pheochromocytoma: a practical guide to clinicians". Current Hypertension Reports. 16 (7): 442. doi:10.1007/s11906-014-0442-z. PMID 24792093. S2CID 38357313.
- Buhl T, Mortensen J, Kjaer A (March 2002). "I-123 MIBG imaging and intraoperative localization of metastatic pheochromocytoma: a case report". Clinical Nuclear Medicine. 27 (3): 183–5. doi:10.1097/00003072-200203000-00007. PMID 11852305. S2CID 11485950.
- De Filpo G, Maggi M, Mannelli M, Canu L (June 2020). "Management and outcome of metastatic pheochromocytomas/paragangliomas: an overview". Journal of Endocrinological Investigation. 44 (1): 15–25. doi:10.1007/s40618-020-01344-z. PMID 32602077. S2CID 220150512.
- Breen W, Bancos I, Young WF, Bible KC, Laack NN, Foote RL, Hallemeier CL (2017-11-22). "External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma". Advances in Radiation Oncology. 3 (1): 25–29. doi:10.1016/j.adro.2017.11.002. PMC 5856976. PMID 29556576.
- Kohlenberg J, Welch B, Hamidi O, Callstrom M, Morris J, Sprung J, et al. (February 2019). "Efficacy and Safety of Ablative Therapy in the Treatment of Patients with Metastatic Pheochromocytoma and Paraganglioma". Cancers. 11 (2): 195. doi:10.3390/cancers11020195. PMC 6407137. PMID 30736463.
- Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EP (November 2014). "Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis". Clinical Endocrinology. 81 (5): 642–51. doi:10.1111/cen.12542. PMID 25041164. S2CID 5407678.
- Averbuch SD, Steakley CS, Young RC, Gelmann EP, Goldstein DS, Stull R, Keiser HR (August 1988). "Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine". Annals of Internal Medicine. 109 (4): 267–73. doi:10.7326/0003-4819-109-4-267. PMID 3395037.
- Huang H, Abraham J, Hung E, Averbuch S, Merino M, Steinberg SM, et al. (October 2008). "Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients". Cancer. 113 (8): 2020–8. doi:10.1002/cncr.23812. PMC 9094399. PMID 18780317. S2CID 205653109.
- Nomura K, Kimura H, Shimizu S, Kodama H, Okamoto T, Obara T, Takano K (August 2009). "Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy". The Journal of Clinical Endocrinology and Metabolism. 94 (8): 2850–6. doi:10.1210/jc.2008-2697. PMID 19470630.
- Jawed I, Velarde M, Därr R, Wolf KI, Adams K, Venkatesan AM, et al. (July 2018). "Continued Tumor Reduction of Metastatic Pheochromocytoma/Paraganglioma Harboring Succinate Dehydrogenase Subunit B Mutations with Cyclical Chemotherapy". Cellular and Molecular Neurobiology. 38 (5): 1099–1106. doi:10.1007/s10571-018-0579-4. PMC 5976545. PMID 29623478.
- Tena I, Gupta G, Tajahuerce M, Benavent M, Cifrián M, Falcon A, et al. (2018). "Successful Second-Line Metronomic Temozolomide in Metastatic Paraganglioma: Case Reports and Review of the Literature". Clinical Medicine Insights: Oncology. 12: 1179554918763367. doi:10.1177/1179554918763367. PMC 5922490. PMID 29720885.
- Tong A, Li M, Cui Y, Ma X, Wang H, Li Y (2020). "Temozolomide Is a Potential Therapeutic Tool for Patients With Metastatic Pheochromocytoma/Paraganglioma-Case Report and Review of the Literature". Frontiers in Endocrinology. 11: 61. doi:10.3389/fendo.2020.00061. PMC 7040234. PMID 32132978.
- Hadoux J, Favier J, Scoazec JY, Leboulleux S, Al Ghuzlan A, Caramella C, et al. (December 2014). "SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma". International Journal of Cancer. 135 (11): 2711–20. doi:10.1002/ijc.28913. PMID 24752622. S2CID 23557293.
- Pryma DA, Chin BB, Noto RB, Dillon JS, Perkins S, Solnes L, et al. (May 2019). "131I-MIBG Therapy in Patients with Advanced Pheochromocytoma or Paraganglioma". Journal of Nuclear Medicine. 60 (5): 623–630. doi:10.2967/jnumed.118.217463. PMC 6495236. PMID 30291194.
- Agrawal A, Rangarajan V, Shah S, Puranik A, Purandare N (November 2018). "MIBG (metaiodobenzylguanidine) theranostics in pediatric and adult malignancies". The British Journal of Radiology. 91 (1091): 20180103. doi:10.1259/bjr.20180103. PMC 6475939. PMID 30048149.
- Carrasquillo JA, Pandit-Taskar N, Chen CC (May 2016). "I-131 Metaiodobenzylguanidine Therapy of Pheochromocytoma and Paraganglioma". Seminars in Nuclear Medicine. 46 (3): 203–14. doi:10.1053/j.semnuclmed.2016.01.011. PMID 27067501.
- Kong G, Grozinsky-Glasberg S, Hofman MS, Callahan J, Meirovitz A, Maimon O, et al. (September 2017). "Efficacy of Peptide Receptor Radionuclide Therapy for Functional Metastatic Paraganglioma and Pheochromocytoma". The Journal of Clinical Endocrinology and Metabolism. 102 (9): 3278–3287. doi:10.1210/jc.2017-00816. PMID 28605448. S2CID 3759391.
- Strosberg J, Wolin E, Chasen B, Kulke M, Bushnell D, Caplin M, et al. (September 2018). "177Lu-Dotatate in the Phase III NETTER-1 Trial". Journal of Clinical Oncology. 36 (25): 2578–2584. doi:10.1200/JCO.2018.78.5865. PMC 6366953. PMID 29878866.
- Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. (January 2017). "177Lu-Dotatate for Midgut Neuroendocrine Tumors". The New England Journal of Medicine. 376 (2): 125–135. doi:10.1056/NEJMoa1607427. PMC 5895095. PMID 28076709.
- Vyakaranam AR, Crona J, Norlén O, Granberg D, Garske-Román U, Sandström M, et al. (June 2019). "177Lu-DOTATATE". Cancers. 11 (7). doi:10.3390/cancers11070909. PMC 6678507. PMID 31261748.
- Satapathy S, Mittal BR, Bhansali A (December 2019). "Peptide receptor radionuclide therapy in the management of advanced pheochromocytoma and paraganglioma: A systematic review and meta-analysis". Clinical Endocrinology. 91 (6): 718–727. doi:10.1111/cen.14106. PMID 31569282. S2CID 203622655.
- Wolf KI, Jha A, van Berkel A, Wild D, Janssen I, Millo CM, et al. (June 2019). "177Lu-DOTATATE". Nuclear Medicine and Molecular Imaging. 53 (3): 223–230. doi:10.1007/s13139-019-00579-w. PMC 6554376. PMID 31231443.
- "NCI Dictionary of Cancer Terms – National Cancer Institute". www.cancer.gov. 2011-02-02. Retrieved 2020-08-18.
- "Pheochromocytoma – National Cancer Institute". www.cancer.gov. 2020-02-12. Retrieved 2020-08-18.
- Noshiro T, Shimizu K, Watanabe T, Akama H, Shibukawa S, Miura W, et al. (January 2000). "Changes in clinical features and long-term prognosis in patients with pheochromocytoma". American Journal of Hypertension. 13 (1 Pt 1): 35–43. doi:10.1016/S0895-7061(99)00139-9. PMID 10678269.
- Hescot S, Curras-Freixes M, Deutschbein T, van Berkel A, Vezzosi D, Amar L, et al. (June 2019). "Prognosis of Malignant Pheochromocytoma and Paraganglioma (MAPP-Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study". The Journal of Clinical Endocrinology and Metabolism. 104 (6): 2367–2374. doi:10.1210/jc.2018-01968. PMID 30715419.
- Hamidi O (June 2019). "Metastatic pheochromocytoma and paraganglioma: recent advances in prognosis and management". Current Opinion in Endocrinology, Diabetes and Obesity. 26 (3): 146–154. doi:10.1097/med.0000000000000476. PMID 30893083. S2CID 84844032.
- Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, et al. (February 2007). "Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005". Nature Clinical Practice. Endocrinology & Metabolism. 3 (2): 92–102. doi:10.1038/ncpendmet0396. PMID 17237836. S2CID 23952363.
- Chen H, Sippel RS, O'Dorisio MS, Vinik AI, Lloyd RV, Pacak K (August 2010). "The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer". Pancreas. 39 (6): 775–83. doi:10.1097/MPA.0b013e3181ebb4f0. PMC 3419007. PMID 20664475.
- Sutton MG, Sheps SG, Lie JT (June 1981). "Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series". Mayo Clinic Proceedings. 56 (6): 354–60. PMID 6453259.
- Kim JH, Moon H, Noh J, Lee J, Kim SG (March 2020). "Epidemiology and Prognosis of Pheochromocytoma/Paraganglioma in Korea: A Nationwide Study Based on the National Health Insurance Service". Endocrinology and Metabolism. 35 (1): 157–164. doi:10.3803/EnM.2020.35.1.157. PMC 7090309. PMID 32207276.
- Berends AM, Buitenwerf E, de Krijger RR, Veeger NJ, van der Horst-Schrivers AN, Links TP, Kerstens MN (May 2018). "Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: A nationwide study and systematic review". European Journal of Internal Medicine. 51: 68–73. doi:10.1016/j.ejim.2018.01.015. PMID 29361475.
- Ebbehoj AL, Sondergaard E, Trolle C, Stochholm K, Poulsen PL (2017-05-03). "The epidemiology of pheochromocytoma: increasing incidence and changing clinical presentation. A population-based retrospective study 1977–2015". Endocrine Abstracts. doi:10.1530/endoabs.49.oc1.4. ISSN 1479-6848.
- Aygun N, Uludag M (2020-06-03). "Pheochromocytoma and Paraganglioma: From Epidemiology to Clinical Findings". Sisli Etfal Hastanesi Tip Bulteni. 54 (2): 159–168. doi:10.14744/SEMB.2020.18794. PMC 7326683. PMID 32617052.
- Antonio K, Valdez MM, Mercado-Asis L, Taïeb D, Pacak K (February 2020). "Pheochromocytoma/paraganglioma: recent updates in genetics, biochemistry, immunohistochemistry, metabolomics, imaging and therapeutic options". Gland Surgery. 9 (1): 105–123. doi:10.21037/gs.2019.10.25. PMC 7082276. PMID 32206603.
- Conzo G, Pasquali D, Colantuoni V, Circelli L, Tartaglia E, Gambardella C, et al. (2014-05-01). "Current concepts of pheochromocytoma". International Journal of Surgery. 12 (5): 469–74. doi:10.1016/j.ijsu.2014.04.001. PMID 24727002.
- Sugrue C (1800). "A Case of Gastrodynia". Med Phys J. 4: 228–331.
- Bausch B, Tischler AS, Schmid KW, Leijon H, Eng C, Neumann HP (July 2017). "Max Schottelius: Pioneer in Pheochromocytoma". Journal of the Endocrine Society. 1 (7): 957–964. doi:10.1210/js.2017-00208. PMC 5689150. PMID 29264546.
- Fränkel, Felix (February 1886). "Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat und Retinitis". Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin. 103 (2): 244–263. doi:10.1007/bf01938677. ISSN 0945-6317. S2CID 31941439.
- Kantorovich V, Pacak K (2010). "Pheochromocytoma and paraganglioma". Neuroendocrinology - Pathological Situations and Diseases. Progress in Brain Research. Vol. 182. Elsevier. pp. 343–73. doi:10.1016/s0079-6123(10)82015-1. ISBN 978-0-444-53616-7. PMC 4714594. PMID 20541673.
- Kiernan CM, Solórzano CC (January 2016). "Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, and Treatment". Surgical Oncology Clinics of North America. 25 (1): 119–38. doi:10.1016/j.soc.2015.08.006. PMID 26610778.
- Welbourn RB (July 1987). "Early surgical history of phaeochromocytoma". The British Journal of Surgery. 74 (7): 594–6. doi:10.1002/bjs.1800740717. PMID 3304519. S2CID 40507310.
- Jacob M, Macwana S, Vivekanand D (March 2015). "Anaesthetic management of a case of adrenal and extra-adrenal phaeochromocytoma for preoperative embolisation". Indian Journal of Anaesthesia. 59 (3): 196–7. doi:10.4103/0019-5049.153046. PMC 4378085. PMID 25838596.
- Boening, Andreas; Burger, Heiko (January 2018). "If You Hear Hoof Beats, Think Horses, Not Zebras". The Thoracic and Cardiovascular Surgeon Reports. 7 (1): e35. doi:10.1055/s-0038-1660808. ISSN 2194-7635. PMC 6033608. PMID 29984129.
- "Rare Disease Day 2021 – 28 Feb". Rare Disease Day – 28 Feb 2021. Retrieved 2020-08-26.
- "Home". NORD (National Organization for Rare Disorders). Retrieved 2020-08-26.
- Sanders, Lisa (2019-10-30). "Why Did the Young Mother Have Searing Head Pain and a Racing Heart?". The New York Times. ISSN 0362-4331. Retrieved 2020-08-26.
- "Neuroendocrine Cancer Survivor Featured on Discovery Fit & Health TV Show". Carcinoid Cancer Foundation. 2012-07-08. Retrieved 2020-08-26.
- "VHL Family Alliance Applauds Grey's Anatomy for Featuring von Hippel-Lindau Disease". www.prnewswire.com. Retrieved 2020-08-26.
- Kevin (2011-01-12). "Mischaracterizations by the popular media of medical conditions". KevinMD.com. Retrieved 2020-08-26.
- "'Grey's Anatomy' Features Rare Disease on Three-Episode Series". ABC News. Retrieved 2020-08-26.
Further reading
- Pourian M, Mostafazadeh DB, Soltani A (2015). "Does this patient have pheochromocytoma? A systematic review of clinical signs and symptoms". Journal of Diabetes and Metabolic Disorders. 15: 11. doi:10.1186/s40200-016-0230-1. PMC 4815191. PMID 27034920.
External links
- "General Information About Pheochromocytoma and Paraganglioma" from the National Cancer Institute
- Pheochromocytoma and Paraganglioma from the American Society of Clinical Oncology
- Pheochromocytoma; Rare Disease Database from the National Organization for Rare Disorders
- What's to Know about Pheochromocytoma from Medical News Today
- MedlinePlus Overview pheochromocytoma
- GeneReviews entry on "Hereditary Paraganglioma-Pheochromocytoma Syndromes"