Pyrrole
Pyrrole is a heterocyclic aromatic organic compound, a five-membered ring with the formula C4H4NH.[3] It is a colorless volatile liquid that darkens readily upon exposure to air. Substituted derivatives are also called pyrroles, e.g., N-methylpyrrole, C4H4NCH3. Porphobilinogen, a trisubstituted pyrrole, is the biosynthetic precursor to many natural products such as heme.[4]
| |||
| |||
| Names | |||
|---|---|---|---|
| Preferred IUPAC name
1H-Pyrrole[1] | |||
Other names
| |||
| Identifiers | |||
3D model (JSmol) |
|||
Beilstein Reference |
1159 | ||
| ChEBI | |||
| ChEMBL | |||
| ChemSpider | |||
| ECHA InfoCard | 100.003.387 | ||
| EC Number |
| ||
Gmelin Reference |
1705 | ||
PubChem CID |
|||
| RTECS number |
| ||
| UNII | |||
| UN number | 1992, 1993 | ||
CompTox Dashboard (EPA) |
|||
| |||
| |||
| Properties | |||
| C4H5N | |||
| Molar mass | 67.091 g·mol−1 | ||
| Density | 0.967 g cm−3 | ||
| Melting point | −23 °C (−9 °F; 250 K) | ||
| Boiling point | 129 to 131 °C (264 to 268 °F; 402 to 404 K) | ||
| Vapor pressure | 7 mmHg at 23 °C | ||
| Acidity (pKa) | 16.5 (for the N−H proton) | ||
| Basicity (pKb) | 13.6 (pKa 0.4 for C.A.) | ||
| −47.6×10−6 cm3 mol−1 | |||
| Viscosity | 0.001225 Pa s | ||
| Thermochemistry | |||
Heat capacity (C) |
1.903 J K−1 mol−1 | ||
Std enthalpy of formation (ΔfH⦵298) |
108.2 kJ mol−1 (gas) | ||
Std enthalpy of combustion (ΔcH⦵298) |
2242 kJ mol−1 | ||
| Hazards | |||
| NFPA 704 (fire diamond) | |||
| Flash point | 33.33 °C (91.99 °F; 306.48 K) | ||
Autoignition temperature |
550 °C (1,022 °F; 823 K) | ||
| Explosive limits | 3.1–14.8% | ||
| Safety data sheet (SDS) | Chemical Safety Data | ||
| Related compounds | |||
Related compounds |
Phosphole, arsole, bismole, stibole | ||
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |||
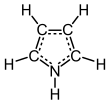
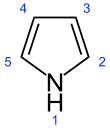

Pyrroles are components of more complex macrocycles, including the porphyrinogens and products derived therefrom, including porphyrins of heme, the chlorins, bacteriochlorins, and chlorophylls.[5]
Properties
Pyrrole is a colorless volatile liquid that darkens readily upon exposure to air, and is usually purified by distillation immediately before use.[6] Pyrrole has a nutty odor. Pyrrole is a 5-membered aromatic heterocycle, like furan and thiophene. Unlike furan and thiophene, it has a dipole in which the positive end lies on the side of the heteroatom, with a dipole moment of 1.58 D. In CDCl3, it has chemical shifts at 6.68 (H2, H5) and 6.22 (H3, H4). Pyrrole is an extremely weak base for an amine, with a conjugate acid pKa of −3.8. The most thermodynamically stable pyrrolium cation (C4H6N+) is formed by protonation at the 2 position. Substitution of pyrrole with alkyl substituents provides a more basic molecule—for example, tetramethylpyrrole has a conjugate acid pKa of +3.7. Pyrrole is also weakly acidic at the N–H position, with a pKa of 16.5. As a hydrogen bonding Lewis acid it is classified as a hard acid and the ECW model lists its acid parameters as EA = 1.38 and CA = 0.68.
History
Pyrrole was first detected by F. F. Runge in 1834, as a constituent of coal tar.[7] In 1857, it was isolated from the pyrolysate of bone. Its name comes from the Greek pyrrhos (πυρρός, “reddish, fiery”), from the reaction used to detect it—the red color that it imparts to wood when moistened with hydrochloric acid.[8]
Occurrence in nature
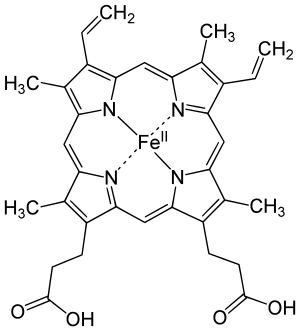
Pyrrole itself is not naturally occurring, but many of its derivatives are found in a variety of cofactors and natural products. Common naturally produced molecules containing pyrroles include vitamin B12, bile pigments like bilirubin and biliverdin, and the porphyrins of heme, chlorophyll, chlorins, bacteriochlorins, and porphyrinogens.[5] Other pyrrole-containing secondary metabolites include PQQ, makaluvamine M, ryanodine, rhazinilam, lamellarin, prodigiosin, myrmicarin, and sceptrin. The syntheses of pyrrole-containing haemin, synthesized by Hans Fischer was recognized by the Nobel Prize.
Pyrrole is a constituent of tobacco smoke and may contribute to its toxic effects.[9]
Synthesis
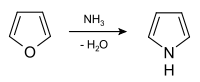
Pyrrole is prepared industrially by treatment of furan with ammonia in the presence of solid acid catalysts, like SiO2 and Al2O3.[8]
Pyrrole can also be formed by catalytic dehydrogenation of pyrrolidine.
Laboratory routes
Several syntheses of the pyrrole ring have been described.[10]
Hantzsch pyrrole synthesis
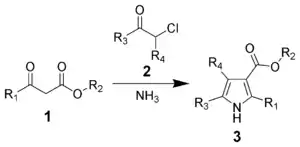
The Hantzsch pyrrole synthesis is the reaction of β-ketoesters (1) with ammonia (or primary amines) and α-haloketones (2) to give substituted pyrroles (3).[11][12]
Knorr pyrrole synthesis
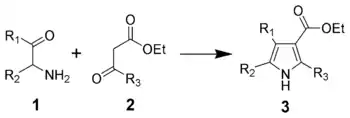
The Knorr pyrrole synthesis involves the reaction of an α-amino ketone or an α-amino-β-ketoester with an activated methylene compound.[13][14][15] The method involves the reaction of an α-aminoketone (1) and a compound containing a methylene group α to (bonded to the next carbon to) a carbonyl group (2).[16]
Paal–Knorr pyrrole synthesis
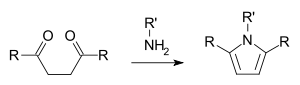
In the Paal–Knorr pyrrole synthesis, a 1,4-dicarbonyl compound reacts with ammonia or a primary amine to form a substituted pyrrole.[17][18]
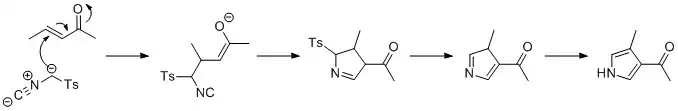
Van Leusen reaction
The Van Leusen reaction can be used to form pyrroles, by reaction of tosylmethyl isocyanide (TosMIC) with an enone in the presence of base, in a Michael addition. A 5-endo cyclization then forms the 5-membered ring, which reacts to eliminate the tosyl group. The last step is tautomerization to the pyrrole.
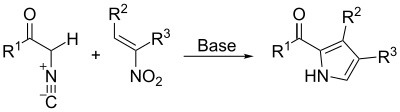
Barton–Zard synthesis
The Barton–Zard synthesis proceeds in a manner similar to the Van Leusen synthesis. An isocyanoacetate reacts with a nitroalkene in a 1,4-addition, followed by 5-endo-dig cyclization, elimination of the nitro group, and tautomerization.[19]
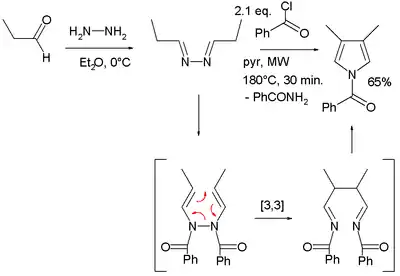
Piloty–Robinson pyrrole synthesis
The starting materials in the Piloty–Robinson pyrrole synthesis, named for Gertrude and Robert Robinson and Oskar Piloty, are two equivalents of an aldehyde and hydrazine.[20][21] The product is a pyrrole with substituents at the 3 and 4 positions. The aldehyde reacts with the diamine to an intermediate di-imine (R−C=N−N=C−R). In the second step, a [3,3]-sigmatropic rearrangement takes place between. Addition of hydrochloric acid leads to ring closure and loss of ammonia to form the pyrrole. The mechanism was developed by the Robinsons.
In one modification, propionaldehyde is treated first with hydrazine and then with benzoyl chloride at high temperatures and assisted by microwave irradiation:[22]
Cycloaddition-based routes
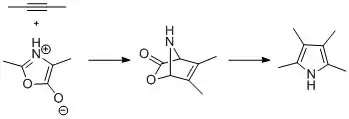
Pyrroles bearing multiple substituents are obtained from the reaction of münchnones and alkynes. The reaction mechanism involves 1,3-dipolar cycloaddition followed by loss of carbon dioxide by a retro-Diels–Alder process. Similar reactions can be performed using azalactones.
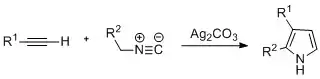
Pyrroles can be prepared by silver-catalyzed cyclization of alkynes with isonitriles, where R2 is an electron-withdrawing group, and R1 is an alkane, aryl group, or ester. Examples of disubstituted alkynes have also been seen to form the desired pyrrole in considerable yield. The reaction is proposed to proceed via a silver acetylide intermediate. This method is analogous to the azide–alkyne click chemistry used to form azoles.
Other methods
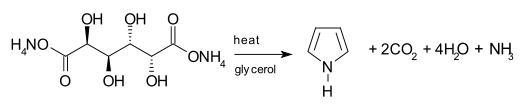
One synthetic route to pyrrole involves the decarboxylation of ammonium mucate, the ammonium salt of mucic acid. The salt is typically heated in a distillation setup with glycerol as a solvent.[23]
Biosynthesis of pyrroles
The de novo biosynthesis of pyrrole rings begins with aminolevulinic acid (ALA), which is synthesized from glycine and succinyl-CoA. ALA dehydratase catalyzes the condensation of two ALA molecules via a Knorr-type ring synthesis to form porphobilinogen (PBG). This later reacts to form, for example, the macrocycles heme and chlorophyll.[24]
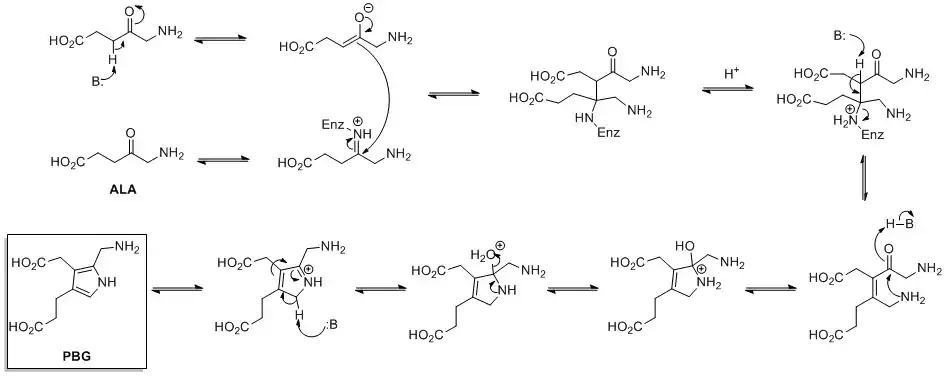
.
Proline is biosynthetically derived from the amino acid L-glutamate. Glutamate-5-semialdehyde is first formed by glutamate 5-kinase (ATP-dependent) and glutamate-5-semialdehyde dehydrogenase (which requires NADH or NADPH). This can then either spontaneously cyclize to form 1-pyrroline-5-carboxylic acid, which is reduced to proline by pyrroline-5-carboxylate reductase (using NADH or NADPH), or turned into ornithine by ornithine aminotransferase, followed by cyclisation by ornithine cyclodeaminase to form proline.[25]
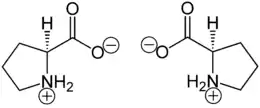
Proline can be used as precursor of aromatic pyrroles in secondary natural products, as in prodigiosins.
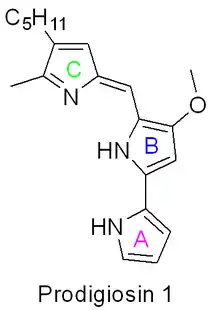
The biosynthesis of Prodigiosin[26][27] involves the convergent coupling of three pyrrole type rings (labeled A, B, and C in figure 1) from L-proline, L-serine, L-methionine, pyruvate, and 2-octenal.
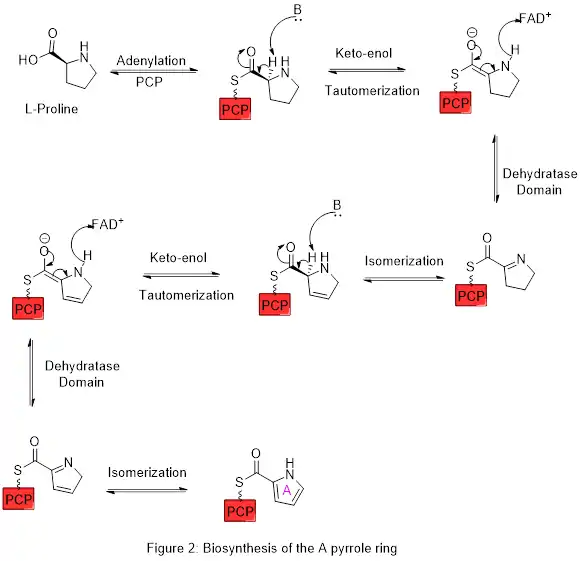
Ring A is synthesized from L-proline through the nonribosomal peptide synthase (NRPS) pathway (figure 2), wherein the pyrrolidine ring of proline is oxidized twice through FAD+ to yield pyrrole ring A.
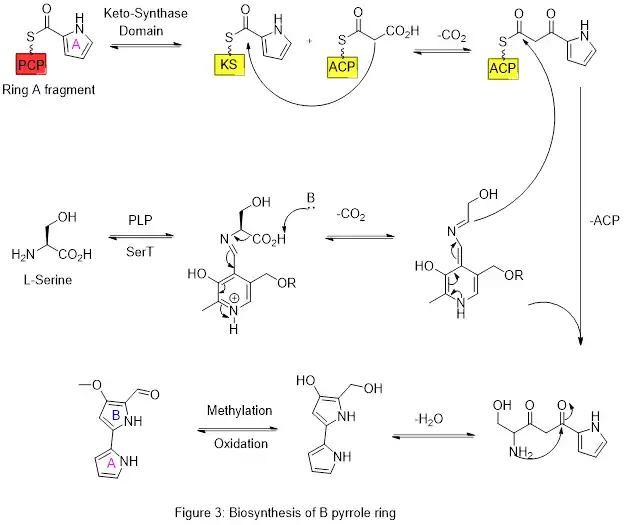
Ring A is then expanded via the polyketide synthase pathway to incorporate L-serine into ring B (figure 3). Ring A fragment is transferred from the peptidyl carrier protein (PCP) to the Acyl Carrier Protein (ACP) by a KS domain, followed by transfer to malonyl-ACP via decarboxylative Claisen condensation. This fragment is then able to react with the masked carbanion formed from the PLP mediated decarboxylation of L-serine, which cyclizes in a dehydration reaction to yield the second pyrrole ring. This intermediate is then modified by methylation (which incorporates a methyl group from L-methionine onto the alcohol at the 6 position) and oxidation of the primary alcohol to the aldehyde to yield the core A–B ring structures.
Reactions and reactivity
Due to its aromatic character, pyrrole is difficult to hydrogenate, does not easily react as a diene in Diels–Alder reactions, and does not undergo usual olefin reactions. Its reactivity is similar to that of benzene and aniline, in that it is easy to alkylate and acylate. Under acidic conditions, pyrroles polymerize easily, and thus many electrophilic reagents that are used in benzene chemistry are not applicable to pyrroles. In contrast, substituted pyrroles (including protected pyrroles) have been used in a broad range of transformations.[10]
Reaction of pyrrole with electrophiles
Pyrroles generally react with electrophiles at the α position (C2 or C5), due to the highest degree of stability of the protonated intermediate.
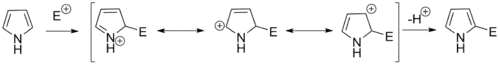
Pyrroles react easily with nitrating (e.g. HNO3/Ac2O), sulfonating (Py·SO3), and halogenating (e.g. NCS, NBS, Br2, SO2Cl2, and KI/H2O2) agents. Halogenation generally provides polyhalogenated pyrroles, but monohalogenation can be performed. As is typical for electrophilic additions to pyrroles, halogenation generally occurs at the 2-position, but can also occur at the 3-position by silation of the nitrogen. This is a useful method for further functionalization of the generally less reactive 3-position.
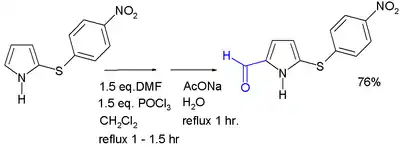
Acylation
Acylation generally occurs at the 2-position, through the use of various methods. Acylation with acid anhydrides and acid chlorides can occur without a catalyst; alternatively, a Lewis acid may be used. 2-Acylpyrroles are also obtained from reaction with nitriles, by the Houben–Hoesch reaction. Pyrrole aldehydes can be formed by a Vilsmeier–Haack reaction. N-Acylation of simple pyrrole does not occur.
Alkylation
Electrophilic alkylation of simple pyrrole is uncommon. Alkylation to form enones at C2 has been seen.
Reaction of deprotonated pyrrole
The NH proton in pyrroles is moderately acidic with a pKa of 16.5. Pyrrole can be deprotonated with strong bases such as butyllithium and sodium hydride. The resulting alkali pyrrolide is nucleophilic. Treating this conjugate base with an electrophile such as iodomethane gives N-methylpyrrole. N-Metalated pyrrole can react with electrophiles at the N or C positions, depending on the coordinating metal. More ionic nitrogen–metal bonds (such as with lithium, sodium, and potassium) and more solvating solvents lead to N-alkylation. Nitrophilic metals, such as MgX, lead to alkylation at C (mainly C2), due to a higher degree of coordination to the nitrogen atom. In the cases of N-substituted pyrroles, metalation of the carbons is more facile. Alkyl groups can be introduced as electrophiles, or by cross-coupling reactions.
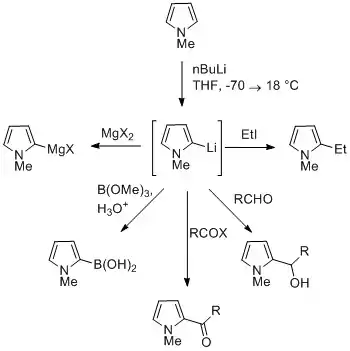
Substitution at C3 can be achieved through the use of N-substituted 3-bromopyrrole, which can be synthesized by bromination of N-silylpyrrole with NBS.
Reductions
Pyrroles can undergo reductions to pyrrolidines and to pyrrolines. For example, Birch reduction of pyrrole esters and amides produced pyrrolines, with the regioselectivity depending on the position of the electron-withdrawing group.
Cyclization reactions
Pyrroles with N-substitution can undergo cycloaddition reactions such as [4+2]-, [2+2]-, and [2+1]-cyclizations. Diels-Alder cyclizations can occur with the pyrrole acting as a diene, especially in the presence of an electron-withdrawing group on the nitrogen. Vinylpyrroles can also act as dienes.
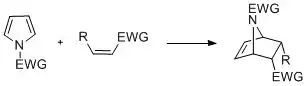
Pyrroles can react with carbenes, such as dichlorocarbene, in a [2+1]-cycloaddition. With dichlorocarbene, a dichlorocyclopropane intermediate is formed, which breaks down to form 3-chloropyridine (the Ciamician–Dennstedt rearrangement).[28][29][30]
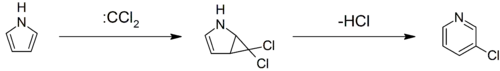
Commercial uses
Polypyrrole is of some commercial value. N-Methylpyrrole is a precursor to N-methylpyrrolecarboxylic acid, a building-block in pharmaceutical chemistry.[8] Pyrroles are also found in several drugs, including atorvastatin, ketorolac, and sunitinib. Pyrroles are used as lightfast red, scarlet, and carmine pigments.[31][32]
Analogs and derivatives
Structural analogs of pyrrole include:
- Pyrroline, a partially saturated analog with one double bond
- Pyrrolidine, the saturated hydrogenated analog
Derivatives of pyrrole include indole, a derivative with a fused benzene ring.
See also
- Simple aromatic rings
- Tetrapyrrole
- Polypyrrole
- Azonine
References
- International Union of Pure and Applied Chemistry (2014). Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013. The Royal Society of Chemistry. p. 141. doi:10.1039/9781849733069. ISBN 978-0-85404-182-4.
- William M. Haynes (2016). CRC Handbook of Chemistry and Physics (97th ed.). Boca Raton: CRC Press. pp. 3–478. ISBN 978-1-4987-5429-3.
- Loudon, Marc G. (2002). "Chemistry of Naphthalene and the Aromatic Heterocycles". Organic Chemistry (4th ed.). New York: Oxford University Press. pp. 1135–1136. ISBN 978-0-19-511999-2.
- Cox, Michael; Lehninger, Albert L.; Nelson, David R. (2000). Lehninger Principles of Biochemistry. New York: Worth Publishers. ISBN 978-1-57259-153-0.
- Jusélius, Jonas; Sundholm, Dage (2000). "The aromatic pathways of porphins, chlorins and bacteriochlorins". Phys. Chem. Chem. Phys. 2 (10): 2145–2151. Bibcode:2000PCCP....2.2145J. doi:10.1039/b000260g.

- Armarego, Wilfred L. F.; Chai, Christina L. L. (2003). Purification of Laboratory Chemicals (5th ed.). Elsevier. p. 346.
- Runge, F. F. (1834). "Ueber einige Produkte der Steinkohlendestillation" [On some products of coal distillation]. Annalen der Physik und Chemie. 31 (5): 65–78. Bibcode:1834AnP...107...65R. doi:10.1002/andp.18341070502. See especially pages 67–68, where Runge names the compound Pyrrol (fire oil) or Rothöl (red oil).
- Harreus, Albrecht Ludwig. "Pyrrole". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a22_453.
- Fowles, Jefferson; Bates, Michael; Noiton, Dominique (March 2000). "The Chemical Constituents in Cigarettes and Cigarette Smoke: Priorities for Harm Reduction" (PDF). Porirua, New Zealand: New Zealand Ministry of Health. pp. 20, 49–65. Retrieved 2012-09-23.
- Lubell, W.; Saint-Cyr, D.; Dufour-Gallant, J.; Hopewell, R.; Boutard, N.; Kassem, T.; Dörr, A.; Zelli, R. (2013). "1H-Pyrroles (Update 2013)". Science of Synthesis. 2013 (1): 157–388.
- Hantzsch, A. (1890). "Neue Bildungsweise von Pyrrolderivaten" [New methods of forming pyrrole derivatives]. Berichte der Deutschen Chemischen Gesellschaft. 23: 1474–1476. doi:10.1002/cber.189002301243.
- Feist, Franz (1902). "Studien in der Furan- und Pyrrol-Gruppe" [Studies in the furan and pyrrole groups]. Berichte der Deutschen Chemischen Gesellschaft. 35 (2): 1537–1544. doi:10.1002/cber.19020350263.
- Knorr, Ludwig (1884). "Synthese von Pyrrolderivaten" [Synthesis of pyrrole derivatives]. Berichte der Deutschen Chemischen Gesellschaft. 17 (2): 1635–1642. doi:10.1002/cber.18840170220.
- Knorr, L. (1886). "Synthetische Versuche mit dem Acetessigester" [Synthesis experiments with the [ethyl] ester of acetoacetic acid]. Annalen der Chemie. 236 (3): 290–332. doi:10.1002/jlac.18862360303.
- Knorr, L.; Lange, H. (1902). "Ueber die Bildung von Pyrrolderivaten aus Isonitrosoketonen" [On the formation of pyrrole derivatives from isonitrosketones]. Berichte der Deutschen Chemischen Gesellschaft. 35 (3): 2998–3008. doi:10.1002/cber.19020350392.
- Corwin, Alsoph Henry (1950). "Chapter 6: The Chemistry of Pyrrole and its Derivatives". In Elderfield, Robert Cooley (ed.). Heterocyclic Compounds. Vol. 1. New York, NY: Wiley. p. 287.
- Paal, C. (1884), "Ueber die Derivate des Acetophenonacetessigesters und des Acetonylacetessigesters", Berichte der Deutschen Chemischen Gesellschaft, 17 (2): 2756–2767, doi:10.1002/cber.188401702228
- Knorr, Ludwig (1884), "Synthese von Furfuranderivaten aus dem Diacetbernsteinsäureester" [Synthesis of furan derivatives from the [diethyl] ester of 2,3-diacetyl-succinic acid], Berichte der Deutschen Chemischen Gesellschaft, 17 (2): 2863–2870, doi:10.1002/cber.188401702254
- Li, Jie Jack (2013). Heterocyclic Chemistry in Drug Discovery. New York: Wiley. ISBN 9781118354421.
- Piloty, Oskar (1910). "Synthese von Pyrrolderivaten: Pyrrole aus Succinylobernsteinsäureester, Pyrrole aus Azinen" [Synthesis of pyrrole derivatives: pyrrole from diethyl succinyl succinate, pyrrole from azines]. Berichte der Deutschen Chemischen Gesellschaft. 43 (1): 489–498. doi:10.1002/cber.19100430182.
- Robinson, Gertrude Maud; Robinson, Robert (1918). "LIV. A new synthesis of tetraphenylpyrrole". J. Chem. Soc. 113: 639–645. doi:10.1039/CT9181300639.
- Milgram, Benjamin C.; Eskildsen, Katrine; Richter, Steven M.; Scheidt, W. Robert; Scheidt, Karl A. (2007). "Microwave-Assisted Piloty–Robinson Synthesis of 3,4-Disubstituted Pyrroles" (Note). J. Org. Chem. 72 (10): 3941–3944. doi:10.1021/jo070389+. PMC 1939979. PMID 17432915.
- Vogel (1956). Practical Organic Chemistry (PDF). p. 837.
- Walsh, Christopher T.; Garneau-Tsodikova, Sylvie; Howard-Jones, Annaleise R. (2006). "Biological formation of pyrroles: Nature's logic and enzymatic machinery". Natural Product Reports. 23 (4): 517–531. doi:10.1039/b605245m. PMID 16874387.
- Lehninger, Albert L.; Nelson, David L.; Cox, Michael M. (2000). Principles of Biochemistry (3rd ed.). New York: W. H. Freeman. ISBN 1-57259-153-6..
- Walsh, C. T.; Garneau-Tsodikova, S.; Howard-Jones, A. R. (2006). "Biological formation of pyrroles: Nature's logic and enzymatic machinery". Nat. Prod. Rep. 23 (4): 517–531. doi:10.1039/b605245m. PMID 16874387.
- Hu, Dennis X. (2016). "Structure, Chemical Synthesis, and Biosynthesis of Prodiginine Natural Products". Chemical Reviews. 116 (14): 7818–7853. doi:10.1021/acs.chemrev.6b00024. PMC 5555159. PMID 27314508.
- Ciamician, G. L.; Dennstedt, M. (1881). "Ueber die Einwirkung des Chloroforms auf die Kaliumverbindung Pyrrols" [On the reaction of chloroform with the potassium compound of pyrrole]. Berichte der Deutschen Chemischen Gesellschaft. 14: 1153–1162. doi:10.1002/cber.188101401240.
- Corwin, Alsoph Henry (1950). Elderfield, Robert Cooley (ed.). Heterocyclic Compounds. Vol. 1. New York, NY: Wiley. p. 309.
- Mosher, H. S. (1950). Elderfield, Robert Cooley (ed.). Heterocyclic Compounds. Vol. 1. New York, NY: Wiley. p. 475.
- "DPP Pigments,Diketopyrrolopyrrole Pigments,DPP Pigments Wholesaler,Diketopyrrolopyrrole Pigments Suppliers". dyes-pigments.standardcon.com.
- Kaur, Matinder; Choi, Dong Hoon (2015). "Diketopyrrolopyrrole: brilliant red pigment dye-based fluorescent probes and their applications". Chemical Society Reviews. 44 (1): 58–77. doi:10.1039/C4CS00248B. PMID 25186723.
Further reading
- Jones, R. Jones, ed. (1990). Pyrroles. Part I. The Synthesis and the Physical and Chemical Aspects of the Pyrrole Ring. Recueil des Travaux Chimiques des Pays-Bas. The Chemistry of Heterocyclic Compounds. Vol. 48. Chichester: John Wiley & Sons. p. 351. doi:10.1002/recl.19911100712. ISBN 978-0-471-62753-1.
- Jolicoeur, Benoit; Chapman, Erin E.; Thompson, Alison; Lubell, William D. (2006). "Pyrrole protection". Tetrahedron. 62 (50): 11531–11563. doi:10.1016/j.tet.2006.08.071.