Protein domain
In molecular biology, a protein domain is a region of a protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains, and a domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length.[1] The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.
Background
The concept of the domain was first proposed in 1973 by Wetlaufer after X-ray crystallographic studies of hen lysozyme[2] and papain[3] and by limited proteolysis studies of immunoglobulins.[4][5] Wetlaufer defined domains as stable units of protein structure that could fold autonomously. In the past domains have been described as units of:
Each definition is valid and will often overlap, i.e. a compact structural domain that is found amongst diverse proteins is likely to fold independently within its structural environment. Nature often brings several domains together to form multidomain and multifunctional proteins with a vast number of possibilities.[9] In a multidomain protein, each domain may fulfill its own function independently, or in a concerted manner with its neighbours. Domains can either serve as modules for building up large assemblies such as virus particles or muscle fibres, or can provide specific catalytic or binding sites as found in enzymes or regulatory proteins.
Example: Pyruvate kinase
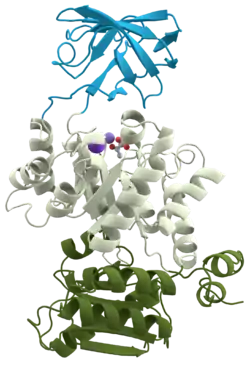
An appropriate example is pyruvate kinase (see first figure), a glycolytic enzyme that plays an important role in regulating the flux from fructose-1,6-biphosphate to pyruvate. It contains an all-β nucleotide binding domain (in blue), an α/β-substrate binding domain (in grey) and an α/β-regulatory domain (in olive green),[10] connected by several polypeptide linkers.[11] Each domain in this protein occurs in diverse sets of protein families.[12]
The central α/β-barrel substrate binding domain is one of the most common enzyme folds. It is seen in many different enzyme families catalysing completely unrelated reactions.[13] The α/β-barrel is commonly called the TIM barrel named after triose phosphate isomerase, which was the first such structure to be solved.[14] It is currently classified into 26 homologous families in the CATH domain database.[15] The TIM barrel is formed from a sequence of β-α-β motifs closed by the first and last strand hydrogen bonding together, forming an eight stranded barrel. There is debate about the evolutionary origin of this domain. One study has suggested that a single ancestral enzyme could have diverged into several families,[16] while another suggests that a stable TIM-barrel structure has evolved through convergent evolution.[17]
The TIM-barrel in pyruvate kinase is 'discontinuous', meaning that more than one segment of the polypeptide is required to form the domain. This is likely to be the result of the insertion of one domain into another during the protein's evolution. It has been shown from known structures that about a quarter of structural domains are discontinuous.[18][19] The inserted β-barrel regulatory domain is 'continuous', made up of a single stretch of polypeptide.
Units of protein structure
The primary structure (string of amino acids) of a protein ultimately encodes its uniquely folded three-dimensional (3D) conformation.[20] The most important factor governing the folding of a protein into 3D structure is the distribution of polar and non-polar side chains.[21] Folding is driven by the burial of hydrophobic side chains into the interior of the molecule so to avoid contact with the aqueous environment. Generally proteins have a core of hydrophobic residues surrounded by a shell of hydrophilic residues. Since the peptide bonds themselves are polar they are neutralised by hydrogen bonding with each other when in the hydrophobic environment. This gives rise to regions of the polypeptide that form regular 3D structural patterns called secondary structure. There are two main types of secondary structure: α-helices and β-sheets.
Some simple combinations of secondary structure elements have been found to frequently occur in protein structure and are referred to as supersecondary structure or motifs. For example, the β-hairpin motif consists of two adjacent antiparallel β-strands joined by a small loop. It is present in most antiparallel β structures both as an isolated ribbon and as part of more complex β-sheets. Another common super-secondary structure is the β-α-β motif, which is frequently used to connect two parallel β-strands. The central α-helix connects the C-termini of the first strand to the N-termini of the second strand, packing its side chains against the β-sheet and therefore shielding the hydrophobic residues of the β-strands from the surface.
Covalent association of two domains represents a functional and structural advantage since there is an increase in stability when compared with the same structures non-covalently associated.[22] Other, advantages are the protection of intermediates within inter-domain enzymatic clefts that may otherwise be unstable in aqueous environments, and a fixed stoichiometric ratio of the enzymatic activity necessary for a sequential set of reactions.[23]
Structural alignment is an important tool for determining domains.
Tertiary structure
Several motifs pack together to form compact, local, semi-independent units called domains.[6] The overall 3D structure of the polypeptide chain is referred to as the protein's tertiary structure. Domains are the fundamental units of tertiary structure, each domain containing an individual hydrophobic core built from secondary structural units connected by loop regions. The packing of the polypeptide is usually much tighter in the interior than the exterior of the domain producing a solid-like core and a fluid-like surface.[24] Core residues are often conserved in a protein family, whereas the residues in loops are less conserved, unless they are involved in the protein's function. Protein tertiary structure can be divided into four main classes based on the secondary structural content of the domain.[25]
- All-α domains have a domain core built exclusively from α-helices. This class is dominated by small folds, many of which form a simple bundle with helices running up and down.
- All-β domains have a core composed of antiparallel β-sheets, usually two sheets packed against each other. Various patterns can be identified in the arrangement of the strands, often giving rise to the identification of recurring motifs, for example the Greek key motif.[26]
- α+β domains are a mixture of all-α and all-β motifs. Classification of proteins into this class is difficult because of overlaps to the other three classes and therefore is not used in the CATH domain database.[15]
- α/β domains are made from a combination of β-α-β motifs that predominantly form a parallel β-sheet surrounded by amphipathic α-helices. The secondary structures are arranged in layers or barrels.
Limits on size
Domains have limits on size.[27] The size of individual structural domains varies from 36 residues in E-selectin to 692 residues in lipoxygenase-1,[18] but the majority, 90%, have fewer than 200 residues[28] with an average of approximately 100 residues.[29] Very short domains, less than 40 residues, are often stabilised by metal ions or disulfide bonds. Larger domains, greater than 300 residues, are likely to consist of multiple hydrophobic cores.[30]
Quaternary structure
Many proteins have a quaternary structure, which consists of several polypeptide chains that associate into an oligomeric molecule. Each polypeptide chain in such a protein is called a subunit. Hemoglobin, for example, consists of two α and two β subunits. Each of the four chains has an all-α globin fold with a heme pocket.
Domain swapping
Domain swapping is a mechanism for forming oligomeric assemblies.[31] In domain swapping, a secondary or tertiary element of a monomeric protein is replaced by the same element of another protein. Domain swapping can range from secondary structure elements to whole structural domains. It also represents a model of evolution for functional adaptation by oligomerisation, e.g. oligomeric enzymes that have their active site at subunit interfaces.[32]
Domains as evolutionary modules
Nature is a tinkerer and not an inventor,[33] new sequences are adapted from pre-existing sequences rather than invented. Domains are the common material used by nature to generate new sequences; they can be thought of as genetically mobile units, referred to as 'modules'. Often, the C and N termini of domains are close together in space, allowing them to easily be "slotted into" parent structures during the process of evolution. Many domain families are found in all three forms of life, Archaea, Bacteria and Eukarya.[34] Protein modules are a subset of protein domains which are found across a range of different proteins with a particularly versatile structure. Examples can be found among extracellular proteins associated with clotting, fibrinolysis, complement, the extracellular matrix, cell surface adhesion molecules and cytokine receptors.[35] Four concrete examples of widespread protein modules are the following domains: SH2, immunoglobulin, fibronectin type 3 and the kringle.[36]
Molecular evolution gives rise to families of related proteins with similar sequence and structure. However, sequence similarities can be extremely low between proteins that share the same structure. Protein structures may be similar because proteins have diverged from a common ancestor. Alternatively, some folds may be more favored than others as they represent stable arrangements of secondary structures and some proteins may converge towards these folds over the course of evolution. There are currently about 110,000 experimentally determined protein 3D structures deposited within the Protein Data Bank (PDB).[37] However, this set contains many identical or very similar structures. All proteins should be classified to structural families to understand their evolutionary relationships. Structural comparisons are best achieved at the domain level. For this reason many algorithms have been developed to automatically assign domains in proteins with known 3D structure; see 'Domain definition from structural co-ordinates'.
The CATH domain database classifies domains into approximately 800 fold families; ten of these folds are highly populated and are referred to as 'super-folds'. Super-folds are defined as folds for which there are at least three structures without significant sequence similarity.[38] The most populated is the α/β-barrel super-fold, as described previously.
Multidomain proteins
The majority of proteins, two-thirds in unicellular organisms and more than 80% in metazoa, are multidomain proteins.[39] However, other studies concluded that 40% of prokaryotic proteins consist of multiple domains while eukaryotes have approximately 65% multi-domain proteins.[40]
Many domains in eukaryotic multidomain proteins can be found as independent proteins in prokaryotes,[41] suggesting that domains in multidomain proteins have once existed as independent proteins. For example, vertebrates have a multi-enzyme polypeptide containing the GAR synthetase, AIR synthetase and GAR transformylase domains (GARs-AIRs-GARt; GAR: glycinamide ribonucleotide synthetase/transferase; AIR: aminoimidazole ribonucleotide synthetase). In insects, the polypeptide appears as GARs-(AIRs)2-GARt, in yeast GARs-AIRs is encoded separately from GARt, and in bacteria each domain is encoded separately.[42]

Origin
Multidomain proteins are likely to have emerged from selective pressure during evolution to create new functions. Various proteins have diverged from common ancestors by different combinations and associations of domains. Modular units frequently move about, within and between biological systems through mechanisms of genetic shuffling:
- transposition of mobile elements including horizontal transfers (between species);[45]
- gross rearrangements such as inversions, translocations, deletions and duplications;
- homologous recombination;
- slippage of DNA polymerase during replication.
Types of organization
The simplest multidomain organization seen in proteins is that of a single domain repeated in tandem.[46] The domains may interact with each other (domain-domain interaction) or remain isolated, like beads on string. The giant 30,000 residue muscle protein titin comprises about 120 fibronectin-III-type and Ig-type domains.[47] In the serine proteases, a gene duplication event has led to the formation of a two β-barrel domain enzyme.[48] The repeats have diverged so widely that there is no obvious sequence similarity between them. The active site is located at a cleft between the two β-barrel domains, in which functionally important residues are contributed from each domain. Genetically engineered mutants of the chymotrypsin serine protease were shown to have some proteinase activity even though their active site residues were abolished and it has therefore been postulated that the duplication event enhanced the enzyme's activity.[48]
Modules frequently display different connectivity relationships, as illustrated by the kinesins and ABC transporters. The kinesin motor domain can be at either end of a polypeptide chain that includes a coiled-coil region and a cargo domain.[49] ABC transporters are built with up to four domains consisting of two unrelated modules, ATP-binding cassette and an integral membrane module, arranged in various combinations.
Not only do domains recombine, but there are many examples of a domain having been inserted into another. Sequence or structural similarities to other domains demonstrate that homologues of inserted and parent domains can exist independently. An example is that of the 'fingers' inserted into the 'palm' domain within the polymerases of the Pol I family.[50] Since a domain can be inserted into another, there should always be at least one continuous domain in a multidomain protein. This is the main difference between definitions of structural domains and evolutionary/functional domains. An evolutionary domain will be limited to one or two connections between domains, whereas structural domains can have unlimited connections, within a given criterion of the existence of a common core. Several structural domains could be assigned to an evolutionary domain.
A superdomain consists of two or more conserved domains of nominally independent origin, but subsequently inherited as a single structural/functional unit.[51] This combined superdomain can occur in diverse proteins that are not related by gene duplication alone. An example of a superdomain is the protein tyrosine phosphatase–C2 domain pair in PTEN, tensin, auxilin and the membrane protein TPTE2. This superdomain is found in proteins in animals, plants and fungi. A key feature of the PTP-C2 superdomain is amino acid residue conservation in the domain interface.
Domains are autonomous folding units
Folding
Protein folding - the unsolved problem : Since the seminal work of Anfinsen in the early 1960s,[20] the goal to completely understand the mechanism by which a polypeptide rapidly folds into its stable native conformation remains elusive. Many experimental folding studies have contributed much to our understanding, but the principles that govern protein folding are still based on those discovered in the very first studies of folding. Anfinsen showed that the native state of a protein is thermodynamically stable, the conformation being at a global minimum of its free energy.
Folding is a directed search of conformational space allowing the protein to fold on a biologically feasible time scale. The Levinthal paradox states that if an averaged sized protein would sample all possible conformations before finding the one with the lowest energy, the whole process would take billions of years.[52] Proteins typically fold within 0.1 and 1000 seconds. Therefore, the protein folding process must be directed some way through a specific folding pathway. The forces that direct this search are likely to be a combination of local and global influences whose effects are felt at various stages of the reaction.[53]
Advances in experimental and theoretical studies have shown that folding can be viewed in terms of energy landscapes,[54][55] where folding kinetics is considered as a progressive organisation of an ensemble of partially folded structures through which a protein passes on its way to the folded structure. This has been described in terms of a folding funnel, in which an unfolded protein has a large number of conformational states available and there are fewer states available to the folded protein. A funnel implies that for protein folding there is a decrease in energy and loss of entropy with increasing tertiary structure formation. The local roughness of the funnel reflects kinetic traps, corresponding to the accumulation of misfolded intermediates. A folding chain progresses toward lower intra-chain free-energies by increasing its compactness. The chain's conformational options become increasingly narrowed ultimately toward one native structure.
Advantage of domains in protein folding
The organisation of large proteins by structural domains represents an advantage for protein folding, with each domain being able to individually fold, accelerating the folding process and reducing a potentially large combination of residue interactions. Furthermore, given the observed random distribution of hydrophobic residues in proteins,[56] domain formation appears to be the optimal solution for a large protein to bury its hydrophobic residues while keeping the hydrophilic residues at the surface.[57][58]
However, the role of inter-domain interactions in protein folding and in energetics of stabilisation of the native structure, probably differs for each protein. In T4 lysozyme, the influence of one domain on the other is so strong that the entire molecule is resistant to proteolytic cleavage. In this case, folding is a sequential process where the C-terminal domain is required to fold independently in an early step, and the other domain requires the presence of the folded C-terminal domain for folding and stabilisation.[59]
It has been found that the folding of an isolated domain can take place at the same rate or sometimes faster than that of the integrated domain,[60] suggesting that unfavourable interactions with the rest of the protein can occur during folding. Several arguments suggest that the slowest step in the folding of large proteins is the pairing of the folded domains.[30] This is either because the domains are not folded entirely correctly or because the small adjustments required for their interaction are energetically unfavourable,[61] such as the removal of water from the domain interface.
Domains and protein flexibility
Protein domain dynamics play a key role in a multitude of molecular recognition and signaling processes. Protein domains, connected by intrinsically disordered flexible linker domains, induce long-range allostery via protein domain dynamics. The resultant dynamic modes cannot be generally predicted from static structures of either the entire protein or individual domains. They can however be inferred by comparing different structures of a protein (as in Database of Molecular Motions). They can also be suggested by sampling in extensive molecular dynamics trajectories[62] and principal component analysis,[63] or they can be directly observed using spectra[64][65] measured by neutron spin echo spectroscopy.
Domain definition from structural co-ordinates
The importance of domains as structural building blocks and elements of evolution has brought about many automated methods for their identification and classification in proteins of known structure. Automatic procedures for reliable domain assignment is essential for the generation of the domain databases, especially as the number of known protein structures is increasing. Although the boundaries of a domain can be determined by visual inspection, construction of an automated method is not straightforward. Problems occur when faced with domains that are discontinuous or highly associated.[66] The fact that there is no standard definition of what a domain really is has meant that domain assignments have varied enormously, with each researcher using a unique set of criteria.[67]
A structural domain is a compact, globular sub-structure with more interactions within it than with the rest of the protein.[68] Therefore, a structural domain can be determined by two visual characteristics: its compactness and its extent of isolation.[69] Measures of local compactness in proteins have been used in many of the early methods of domain assignment[70][71][72][73] and in several of the more recent methods.[28][74][75][76][77]
Methods
One of the first algorithms[70] used a Cα-Cα distance map together with a hierarchical clustering routine that considered proteins as several small segments, 10 residues in length. The initial segments were clustered one after another based on inter-segment distances; segments with the shortest distances were clustered and considered as single segments thereafter. The stepwise clustering finally included the full protein. Go[73] also exploited the fact that inter-domain distances are normally larger than intra-domain distances; all possible Cα-Cα distances were represented as diagonal plots in which there were distinct patterns for helices, extended strands and combinations of secondary structures.
The method by Sowdhamini and Blundell clusters secondary structures in a protein based on their Cα-Cα distances and identifies domains from the pattern in their dendrograms.[66] As the procedure does not consider the protein as a continuous chain of amino acids there are no problems in treating discontinuous domains. Specific nodes in these dendrograms are identified as tertiary structural clusters of the protein, these include both super-secondary structures and domains. The DOMAK algorithm is used to create the 3Dee domain database.[75] It calculates a 'split value' from the number of each type of contact when the protein is divided arbitrarily into two parts. This split value is large when the two parts of the structure are distinct.
The method of Wodak and Janin[78] was based on the calculated interface areas between two chain segments repeatedly cleaved at various residue positions. Interface areas were calculated by comparing surface areas of the cleaved segments with that of the native structure. Potential domain boundaries can be identified at a site where the interface area was at a minimum. Other methods have used measures of solvent accessibility to calculate compactness.[28][79][80]
The PUU algorithm[19] incorporates a harmonic model used to approximate inter-domain dynamics. The underlying physical concept is that many rigid interactions will occur within each domain and loose interactions will occur between domains. This algorithm is used to define domains in the FSSP domain database.[74]
Swindells (1995) developed a method, DETECTIVE, for identification of domains in protein structures based on the idea that domains have a hydrophobic interior. Deficiencies were found to occur when hydrophobic cores from different domains continue through the interface region.
RigidFinder is a novel method for identification of protein rigid blocks (domains and loops) from two different conformations. Rigid blocks are defined as blocks where all inter residue distances are conserved across conformations.
The method RIBFIND developed by Pandurangan and Topf identifies rigid bodies in protein structures by performing spacial clustering of secondary structural elements in proteins.[81] The RIBFIND rigid bodies have been used to flexibly fit protein structures into cryo electron microscopy density maps.[82]
A general method to identify dynamical domains, that is protein regions that behave approximately as rigid units in the course of structural fluctuations, has been introduced by Potestio et al.[62] and, among other applications was also used to compare the consistency of the dynamics-based domain subdivisions with standard structure-based ones. The method, termed PiSQRD, is publicly available in the form of a webserver.[83] The latter allows users to optimally subdivide single-chain or multimeric proteins into quasi-rigid domains[62][83] based on the collective modes of fluctuation of the system. By default the latter are calculated through an elastic network model;[84] alternatively pre-calculated essential dynamical spaces can be uploaded by the user.
Example domains
- Armadillo repeats: named after the β-catenin-like Armadillo protein of the fruit fly Drosophila melanogaster.
- Basic leucine zipper domain (bZIP domain): found in many DNA-binding eukaryotic proteins. One part of the domain contains a region that mediates sequence-specific DNA-binding properties and the Leucine zipper that is required for the dimerization of two DNA-binding regions. The DNA-binding region comprises a number of basic aminoacids such as arginine and lysine.
- Cadherin repeats: Cadherins function as Ca2+-dependent cell–cell adhesion proteins. Cadherin domains are extracellular regions which mediate cell-to-cell homophilic binding between cadherins on the surface of adjacent cells.
- Death effector domain (DED): allows protein–protein binding by homotypic interactions (DED-DED). Caspase proteases trigger apoptosis via proteolytic cascades. Pro-caspase-8 and pro-caspase-9 bind to specific adaptor molecules via DED domains, which leads to autoactivation of caspases.
- EF hand: a helix-turn-helix structural motif found in each structural domain of the signaling protein calmodulin and in the muscle protein troponin-C.
- Immunoglobulin-like domains: found in proteins of the immunoglobulin superfamily (IgSF).[85] They contain about 70-110 amino acids and are classified into different categories (IgV, IgC1, IgC2 and IgI) according to their size and function. They possess a characteristic fold in which two beta sheets form a "sandwich" that is stabilized by interactions between conserved cysteines and other charged amino acids. They are important for protein–protein interactions in processes of cell adhesion, cell activation, and molecular recognition. These domains are commonly found in molecules with roles in the immune system.
- Phosphotyrosine-binding domain (PTB): PTB domains usually bind to phosphorylated tyrosine residues. They are often found in signal transduction proteins. PTB-domain binding specificity is determined by residues to the amino-terminal side of the phosphotyrosine. Examples: the PTB domains of both SHC and IRS-1 bind to a NPXpY sequence. PTB-containing proteins such as SHC and IRS-1 are important for insulin responses of human cells.
- Pleckstrin homology domain (PH): PH domains bind phosphoinositides with high affinity. Specificity for PtdIns(3)P, PtdIns(4)P, PtdIns(3,4)P2, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 have all been observed. Given the fact that phosphoinositides are sequestered to various cell membranes (due to their long lipophilic tail) the PH domains usually causes recruitment of the protein in question to a membrane where the protein can exert a certain function in cell signalling, cytoskeletal reorganization or membrane trafficking.
- Src homology 2 domain (SH2): SH2 domains are often found in signal transduction proteins. SH2 domains confer binding to phosphorylated tyrosine (pTyr). Named after the phosphotyrosine binding domain of the src viral oncogene, which is itself a tyrosine kinase. See also: SH3 domain.
- Zinc finger DNA-binding domain (ZnF_GATA): ZnF_GATA domain-containing proteins are typically transcription factors that usually bind to the DNA sequence [AT]GATA[AG] of promoters.
Domains of unknown function
A large fraction of domains are of unknown function. A domain of unknown function (DUF) is a protein domain that has no characterized function. These families have been collected together in the Pfam database using the prefix DUF followed by a number, with examples being DUF2992 and DUF1220. There are now over 3,000 DUF families within the Pfam database representing over 20% of known families.[86] Surprisingly, the number of DUFs in Pfam has increased from 20% (in 2010) to 22% (in 2019), mostly due to an increasing number of new genome sequences. Pfam release 32.0 (2019) contained 3,961 DUFs.[87]
See also
- Binding domain
- PANDIT, a biological database covering protein domains
- Pfam: database of protein domains
- Protein
- Protein structure
- Protein structure prediction
- Protein structure prediction software
- Protein superfamily
- Protein tandem repeats
- Protein family
- Protein subfamily
- Short linear motif
- Structural biology
- Structural Classification of Proteins (SCOP)
- CATH Protein Structure Classification database
References
This article incorporates text and figures from George, R. A. (2002) "Predicting Structural Domains in Proteins" Thesis, University College London, which were contributed by its author.
- Xu D, Nussinov R (1 February 1998). "Favorable domain size in proteins". Folding & Design. 3 (1): 11–7. doi:10.1016/S1359-0278(98)00004-2. PMID 9502316.
- Phillips DC (November 1966). "The three-dimensional structure of an enzyme molecule". Scientific American. 215 (5): 78–90. Bibcode:1966SciAm.215e..78P. doi:10.1038/scientificamerican1166-78. PMID 5978599. S2CID 39959172.
- Drenth J, Jansonius JN, Koekoek R, Swen HM, Wolthers BG (June 1968). "Structure of papain". Nature. 218 (5145): 929–32. Bibcode:1968Natur.218..929D. doi:10.1038/218929a0. PMID 5681232. S2CID 4169127.
- Porter RR (May 1973). "Structural studies of immunoglobulins". Science. 180 (4087): 713–6. Bibcode:1973Sci...180..713P. doi:10.1126/science.180.4087.713. PMID 4122075.
- Edelman GM (May 1973). "Antibody structure and molecular immunology". Science. 180 (4088): 830–40. Bibcode:1973Sci...180..830E. doi:10.1126/science.180.4088.830. PMID 4540988.
- Richardson JS (1981). "The anatomy and taxonomy of protein structure". Advances in Protein Chemistry. 34: 167–339. doi:10.1016/S0065-3233(08)60520-3. ISBN 9780120342341. PMID 7020376.
- Bork P (July 1991). "Shuffled domains in extracellular proteins". FEBS Letters. 286 (1–2): 47–54. doi:10.1016/0014-5793(91)80937-X. PMID 1864378. S2CID 22126481.
- Wetlaufer DB (March 1973). "Nucleation, rapid folding, and globular intrachain regions in proteins". Proceedings of the National Academy of Sciences of the United States of America. 70 (3): 697–701. Bibcode:1973PNAS...70..697W. doi:10.1073/pnas.70.3.697. PMC 433338. PMID 4351801.
- Chothia C (June 1992). "Proteins. One thousand families for the molecular biologist". Nature. 357 (6379): 543–4. Bibcode:1992Natur.357..543C. doi:10.1038/357543a0. PMID 1608464. S2CID 4355476.
- Bakszt R, Wernimont A, Allali-Hassani A, Mok MW, Hills T, Hui R, Pizarro JC (September 2010). "The crystal structure of Toxoplasma gondii pyruvate kinase 1". PLOS ONE. 5 (9): e12736. Bibcode:2010PLoSO...512736B. doi:10.1371/journal.pone.0012736. PMC 2939071. PMID 20856875.
- George RA, Heringa J (November 2002). "An analysis of protein domain linkers: their classification and role in protein folding". Protein Engineering. 15 (11): 871–9. doi:10.1093/protein/15.11.871. PMID 12538906.
- "Protein Domains, Domain Assignment, Identification and Classification According to CATH and SCOP Databases". proteinstructures.com. Retrieved 14 October 2018.
- Hegyi H, Gerstein M (April 1999). "The relationship between protein structure and function: a comprehensive survey with application to the yeast genome". Journal of Molecular Biology. 288 (1): 147–64. CiteSeerX 10.1.1.217.9806. doi:10.1006/jmbi.1999.2661. PMID 10329133.
- Banner DW, Bloomer AC, Petsko GA, Phillips DC, Pogson CI, Wilson IA, et al. (June 1975). "Structure of chicken muscle triose phosphate isomerase determined crystallographically at 2.5 angstrom resolution using amino acid sequence data". Nature. 255 (5510): 609–14. Bibcode:1975Natur.255..609B. doi:10.1038/255609a0. PMID 1134550. S2CID 4195346.
- Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM (August 1997). "CATH--a hierarchic classification of protein domain structures". Structure. 5 (8): 1093–108. doi:10.1016/S0969-2126(97)00260-8. PMID 9309224.
- Copley RR, Bork P (November 2000). "Homology among (betaalpha)(8) barrels: implications for the evolution of metabolic pathways". Journal of Molecular Biology. 303 (4): 627–41. doi:10.1006/jmbi.2000.4152. PMID 11054297.
- Lesk AM, Brändén CI, Chothia C (1989). "Structural principles of alpha/beta barrel proteins: the packing of the interior of the sheet". Proteins. 5 (2): 139–48. doi:10.1002/prot.340050208. PMID 2664768. S2CID 15340449.
- Jones S, Stewart M, Michie A, Swindells MB, Orengo C, Thornton JM (February 1998). "Domain assignment for protein structures using a consensus approach: characterization and analysis". Protein Science. 7 (2): 233–42. doi:10.1002/pro.5560070202. PMC 2143930. PMID 9521098.
- Holm L, Sander C (July 1994). "Parser for protein folding units". Proteins. 19 (3): 256–68. doi:10.1002/prot.340190309. PMID 7937738. S2CID 525264.
- Anfinsen CB, Haber E, Sela M, White FH (September 1961). "The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain". Proceedings of the National Academy of Sciences of the United States of America. 47 (9): 1309–14. Bibcode:1961PNAS...47.1309A. doi:10.1073/pnas.47.9.1309. PMC 223141. PMID 13683522.
- Cordes MH, Davidson AR, Sauer RT (February 1996). "Sequence space, folding and protein design". Current Opinion in Structural Biology. 6 (1): 3–10. doi:10.1016/S0959-440X(96)80088-1. PMID 8696970.
- Ghélis C, Yon JM (July 1979). "[Conformational coupling between structural units. A decisive step in the functional structure formation]". Comptes Rendus de l'Académie des Sciences, Série D. 289 (2): 197–9. PMID 117925.
- Ostermeier M, Benkovic SJ (2000). "Evolution of protein function by domain swapping". Evolutionary Protein Design. Adv Protein Chem. Advances in Protein Chemistry. Vol. 55. pp. 29–77. doi:10.1016/s0065-3233(01)55002-0. ISBN 9780120342556. PMID 11050932.
- Zhou Y, Vitkup D, Karplus M (January 1999). "Native proteins are surface-molten solids: application of the Lindemann criterion for the solid versus liquid state". Journal of Molecular Biology. 285 (4): 1371–5. doi:10.1006/jmbi.1998.2374. PMID 9917381. S2CID 8702994.
- Levitt M, Chothia C (June 1976). "Structural patterns in globular proteins". Nature. 261 (5561): 552–8. Bibcode:1976Natur.261..552L. doi:10.1038/261552a0. PMID 934293. S2CID 4154884.
- Hutchinson EG, Thornton JM (April 1993). "The Greek key motif: extraction, classification and analysis". Protein Engineering. 6 (3): 233–45. doi:10.1093/protein/6.3.233. PMID 8506258.
- Savageau MA (March 1986). "Proteins of Escherichia coli come in sizes that are multiples of 14 kDa: domain concepts and evolutionary implications". Proceedings of the National Academy of Sciences of the United States of America. 83 (5): 1198–202. Bibcode:1986PNAS...83.1198S. doi:10.1073/pnas.83.5.1198. PMC 323042. PMID 3513170.
- Islam SA, Luo J, Sternberg MJ (June 1995). "Identification and analysis of domains in proteins". Protein Engineering. 8 (6): 513–25. doi:10.1093/protein/8.6.513. PMID 8532675.
- Wheelan SJ, Marchler-Bauer A, Bryant SH (July 2000). "Domain size distributions can predict domain boundaries". Bioinformatics. 16 (7): 613–8. doi:10.1093/bioinformatics/16.7.613. PMID 11038331.
- Garel, J. (1992). "Folding of large proteins: Multidomain and multisubunit proteins". In Creighton, T. (ed.). Protein Folding (First ed.). New York: W.H. Freeman and Company. pp. 405–454. ISBN 978-0-7167-7027-5.
- Bennett MJ, Schlunegger MP, Eisenberg D (December 1995). "3D domain swapping: a mechanism for oligomer assembly". Protein Science. 4 (12): 2455–68. doi:10.1002/pro.5560041202. PMC 2143041. PMID 8580836.
- Heringa J, Taylor WR (June 1997). "Three-dimensional domain duplication, swapping and stealing". Current Opinion in Structural Biology. 7 (3): 416–21. doi:10.1016/S0959-440X(97)80060-7. PMID 9204285.
- Jacob F (June 1977). "Evolution and tinkering". Science. 196 (4295): 1161–6. Bibcode:1977Sci...196.1161J. doi:10.1126/science.860134. PMID 860134. S2CID 29756896.
- Ren S, Yang G, He Y, Wang Y, Li Y, Chen Z (October 2008). "The conservation pattern of short linear motifs is highly correlated with the function of interacting protein domains". BMC Genomics. 9: 452. doi:10.1186/1471-2164-9-452. PMC 2576256. PMID 18828911.
- Campbell ID, Downing AK (May 1994). "Building protein structure and function from modular units". Trends in Biotechnology. 12 (5): 168–72. doi:10.1016/0167-7799(94)90078-7. PMID 7764899.
- Bruce, Alberts (18 November 2014). Molecular biology of the cell (Sixth ed.). New York, NY. ISBN 9780815344322. OCLC 887605755.
- wwPDB.org. "wwPDB: Worldwide Protein Data Bank". www.pdb.org. Archived from the original on 7 April 2015. Retrieved 25 July 2007.
- Orengo CA, Jones DT, Thornton JM (December 1994). "Protein superfamilies and domain superfolds". Nature. 372 (6507): 631–4. Bibcode:1994Natur.372..631O. doi:10.1038/372631a0. PMID 7990952. S2CID 4330359.
- Apic G, Gough J, Teichmann SA (July 2001). "Domain combinations in archaeal, eubacterial and eukaryotic proteomes". Journal of Molecular Biology. 310 (2): 311–25. doi:10.1006/jmbi.2001.4776. PMID 11428892. S2CID 11894663.
- Ekman D, Björklund AK, Frey-Skött J, Elofsson A (April 2005). "Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions". Journal of Molecular Biology. 348 (1): 231–43. doi:10.1016/j.jmb.2005.02.007. PMID 15808866.
- Davidson JN, Chen KC, Jamison RS, Musmanno LA, Kern CB (March 1993). "The evolutionary history of the first three enzymes in pyrimidine biosynthesis". BioEssays. 15 (3): 157–64. doi:10.1002/bies.950150303. PMID 8098212. S2CID 24897614.
- Henikoff S, Greene EA, Pietrokovski S, Bork P, Attwood TK, Hood L (October 1997). "Gene families: the taxonomy of protein paralogs and chimeras". Science. 278 (5338): 609–14. Bibcode:1997Sci...278..609H. CiteSeerX 10.1.1.562.2262. doi:10.1126/science.278.5338.609. PMID 9381171.
- Walker WP, Aradhya S, Hu CL, Shen S, Zhang W, Azarani A, et al. (December 2007). "Genetic analysis of attractin homologs". Genesis. 45 (12): 744–56. doi:10.1002/dvg.20351. PMID 18064672. S2CID 20878849.
- "SMART: Main page". smart.embl.de. Retrieved 1 January 2017.
- Bork P, Doolittle RF (October 1992). "Proposed acquisition of an animal protein domain by bacteria". Proceedings of the National Academy of Sciences of the United States of America. 89 (19): 8990–4. Bibcode:1992PNAS...89.8990B. doi:10.1073/pnas.89.19.8990. PMC 50050. PMID 1409594.
- Heringa J (June 1998). "Detection of internal repeats: how common are they?". Current Opinion in Structural Biology. 8 (3): 338–45. doi:10.1016/S0959-440X(98)80068-7. PMID 9666330.
- Politou AS, Gautel M, Improta S, Vangelista L, Pastore A (February 1996). "The elastic I-band region of titin is assembled in a "modular" fashion by weakly interacting Ig-like domains". Journal of Molecular Biology. 255 (4): 604–16. doi:10.1006/jmbi.1996.0050. PMID 8568900.
- McLachlan AD (February 1979). "Gene duplications in the structural evolution of chymotrypsin". Journal of Molecular Biology. 128 (1): 49–79. doi:10.1016/0022-2836(79)90308-5. PMID 430571.
- Moore JD, Endow SA (March 1996). "Kinesin proteins: a phylum of motors for microtubule-based motility". BioEssays. 18 (3): 207–19. doi:10.1002/bies.950180308. PMID 8867735. S2CID 46012215.
- Russell RB (December 1994). "Domain insertion". Protein Engineering. 7 (12): 1407–10. doi:10.1093/protein/7.12.1407. PMID 7716150.
- Haynie DT, Xue B (May 2015). "Superdomains in the protein structure hierarchy: The case of PTP-C2". Protein Science. 24 (5): 874–82. doi:10.1002/pro.2664. PMC 4420535. PMID 25694109.
- Levinthal C (1968). "Are there pathways for protein folding?" (PDF). J Chim Phys. 65: 44–45. Bibcode:1968JCP....65...44L. doi:10.1051/jcp/1968650044. Archived from the original (PDF) on 2 September 2009.
- Dill KA (June 1999). "Polymer principles and protein folding". Protein Science. 8 (6): 1166–80. doi:10.1110/ps.8.6.1166. PMC 2144345. PMID 10386867.
- Leopold PE, Montal M, Onuchic JN (September 1992). "Protein folding funnels: a kinetic approach to the sequence-structure relationship". Proceedings of the National Academy of Sciences of the United States of America. 89 (18): 8721–5. Bibcode:1992PNAS...89.8721L. doi:10.1073/pnas.89.18.8721. PMC 49992. PMID 1528885.
- Dill KA, Chan HS (January 1997). "From Levinthal to pathways to funnels". Nature Structural Biology. 4 (1): 10–9. doi:10.1038/nsb0197-10. PMID 8989315. S2CID 11557990.
- White SH, Jacobs RE (April 1990). "Statistical distribution of hydrophobic residues along the length of protein chains. Implications for protein folding and evolution". Biophysical Journal. 57 (4): 911–21. Bibcode:1990BpJ....57..911W. doi:10.1016/S0006-3495(90)82611-4. PMC 1280792. PMID 2188687.
- George RA, Heringa J (February 2002). "SnapDRAGON: a method to delineate protein structural domains from sequence data". Journal of Molecular Biology. 316 (3): 839–51. CiteSeerX 10.1.1.329.2921. doi:10.1006/jmbi.2001.5387. PMID 11866536.
- George RA, Lin K, Heringa J (July 2005). "Scooby-domain: prediction of globular domains in protein sequence". Nucleic Acids Research. 33 (Web Server issue): W160-3. doi:10.1093/nar/gki381. PMC 1160142. PMID 15980446.
- Desmadril M, Yon JM (July 1981). "Existence of intermediates in the refolding of T4 lysozyme at pH 7.4". Biochemical and Biophysical Research Communications. 101 (2): 563–9. doi:10.1016/0006-291X(81)91296-1. PMID 7306096.
- Teale JM, Benjamin DC (July 1977). "Antibody as immunological probe for studying refolding of bovine serum albumin. Refolding within each domain". The Journal of Biological Chemistry. 252 (13): 4521–6. doi:10.1016/S0021-9258(17)40192-X. PMID 873903.
- Creighton, T. E. (1983). Proteins: Structures and molecular properties. Freeman, New York. Second edition.
- Potestio R, Pontiggia F, Micheletti C (June 2009). "Coarse-grained description of protein internal dynamics: an optimal strategy for decomposing proteins in rigid subunits". Biophysical Journal. 96 (12): 4993–5002. Bibcode:2009BpJ....96.4993P. doi:10.1016/j.bpj.2009.03.051. PMC 2712024. PMID 19527659.
- Baron R, Vellore NA (July 2012). "LSD1/CoREST is an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition". Proceedings of the National Academy of Sciences of the United States of America. 109 (31): 12509–14. Bibcode:2012PNAS..10912509B. doi:10.1073/pnas.1207892109. PMC 3411975. PMID 22802671.
- Farago B, Li J, Cornilescu G, Callaway DJ, Bu Z (November 2010). "Activation of nanoscale allosteric protein domain motion revealed by neutron spin echo spectroscopy". Biophysical Journal. 99 (10): 3473–82. Bibcode:2010BpJ....99.3473F. doi:10.1016/j.bpj.2010.09.058. PMC 2980739. PMID 21081097.
- Bu Z, Biehl R, Monkenbusch M, Richter D, Callaway DJ (December 2005). "Coupled protein domain motion in Taq polymerase revealed by neutron spin-echo spectroscopy". Proceedings of the National Academy of Sciences of the United States of America. 102 (49): 17646–51. Bibcode:2005PNAS..10217646B. doi:10.1073/pnas.0503388102. PMC 1345721. PMID 16306270.
- Sowdhamini R, Blundell TL (March 1995). "An automatic method involving cluster analysis of secondary structures for the identification of domains in proteins". Protein Science. 4 (3): 506–20. doi:10.1002/pro.5560040317. PMC 2143076. PMID 7795532.
- Swindells MB (January 1995). "A procedure for detecting structural domains in proteins". Protein Science. 4 (1): 103–12. doi:10.1002/pro.5560040113. PMC 2142966. PMID 7773168.
- Janin J, Wodak SJ (1983). "Structural domains in proteins and their role in the dynamics of protein function". Progress in Biophysics and Molecular Biology. 42 (1): 21–78. doi:10.1016/0079-6107(83)90003-2. PMID 6353481.
- Tsai CJ, Nussinov R (January 1997). "Hydrophobic folding units derived from dissimilar monomer structures and their interactions". Protein Science. 6 (1): 24–42. doi:10.1002/pro.5560060104. PMC 2143523. PMID 9007974.
- Crippen GM (December 1978). "The tree structural organization of proteins". Journal of Molecular Biology. 126 (3): 315–32. doi:10.1016/0022-2836(78)90043-8. PMID 745231.
- Rossmann MG, Moras D, Olsen KW (July 1974). "Chemical and biological evolution of nucleotide-binding protein". Nature. 250 (463): 194–9. Bibcode:1974Natur.250..194R. doi:10.1038/250194a0. PMID 4368490. S2CID 4273028.
- Rose GD (November 1979). "Hierarchic organization of domains in globular proteins". Journal of Molecular Biology. 134 (3): 447–70. doi:10.1016/0022-2836(79)90363-2. PMID 537072.
- Go N, Taketomi H (February 1978). "Respective roles of short- and long-range interactions in protein folding". Proceedings of the National Academy of Sciences of the United States of America. 75 (2): 559–63. Bibcode:1978PNAS...75..559G. doi:10.1073/pnas.75.2.559. PMC 411294. PMID 273218.
- Holm L, Sander C (January 1997). "Dali/FSSP classification of three-dimensional protein folds". Nucleic Acids Research. 25 (1): 231–4. doi:10.1093/nar/25.1.231. PMC 146389. PMID 9016542.
- Siddiqui AS, Barton GJ (May 1995). "Continuous and discontinuous domains: an algorithm for the automatic generation of reliable protein domain definitions". Protein Science. 4 (5): 872–84. doi:10.1002/pro.5560040507. PMC 2143117. PMID 7663343.
- Zehfus MH (June 1997). "Identification of compact, hydrophobically stabilized domains and modules containing multiple peptide chains". Protein Science. 6 (6): 1210–9. doi:10.1002/pro.5560060609. PMC 2143719. PMID 9194181.
- Taylor WR (March 1999). "Protein structural domain identification". Protein Engineering. 12 (3): 203–16. doi:10.1093/protein/12.3.203. PMID 10235621.
- Wodak SJ, Janin J (November 1981). "Location of structural domains in protein". Biochemistry. 20 (23): 6544–52. doi:10.1021/bi00526a005. PMID 7306523.
- Rashin, 1985
- Zehfus MH, Rose GD (September 1986). "Compact units in proteins". Biochemistry. 25 (19): 5759–65. doi:10.1021/bi00367a062. PMID 3778881.
- Pandurangan AP, Topf M (September 2012). "RIBFIND: a web server for identifying rigid bodies in protein structures and to aid flexible fitting into cryo EM maps" (PDF). Bioinformatics. 28 (18): 2391–3. doi:10.1093/bioinformatics/bts446. PMID 22796953.
- Pandurangan AP, Topf M (February 2012). "Finding rigid bodies in protein structures: Application to flexible fitting into cryoEM maps". Journal of Structural Biology. 177 (2): 520–31. doi:10.1016/j.jsb.2011.10.011. PMID 22079400.
- Aleksiev T, Potestio R, Pontiggia F, Cozzini S, Micheletti C (October 2009). "PiSQRD: a web server for decomposing proteins into quasi-rigid dynamical domains". Bioinformatics. 25 (20): 2743–4. doi:10.1093/bioinformatics/btp512. PMID 19696046. S2CID 28106759.
- Micheletti, C., Carloni, P. and Maritan, A. Accurate and efficient description of protein vibrational dynamics: comparing molecular dynamics and gaussian models, Proteins, 55, 635, 2004.
- Barclay AN (August 2003). "Membrane proteins with immunoglobulin-like domains--a master superfamily of interaction molecules". Seminars in Immunology. 15 (4): 215–23. doi:10.1016/S1044-5323(03)00047-2. PMID 14690046.
- Bateman A, Coggill P, Finn RD (October 2010). "DUFs: families in search of function". Acta Crystallographica. Section F, Structural Biology and Crystallization Communications. 66 (Pt 10): 1148–52. doi:10.1107/S1744309110001685. PMC 2954198. PMID 20944204.
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. (January 2019). "The Pfam protein families database in 2019". Nucleic Acids Research. 47 (D1): D427–D432. doi:10.1093/nar/gky995. PMC 6324024. PMID 30357350.
Key papers
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. (January 2000). "The Protein Data Bank". Nucleic Acids Research. 28 (1): 235–42. doi:10.1093/nar/28.1.235. PMC 102472. PMID 10592235.
- Tooze J, Brändén C (1999). Introduction to protein structure. New York: Garland Pub. ISBN 978-0-8153-2305-1.
- Das S, Smith TF (2000). "Identifying nature's protein Lego set". Advances in Protein Chemistry. 54: 159–83. doi:10.1016/S0065-3233(00)54006-6. ISBN 978-0-12-034254-9. PMID 10829228.
- Dietmann S, Park J, Notredame C, Heger A, Lappe M, Holm L (January 2001). "A fully automatic evolutionary classification of protein folds: Dali Domain Dictionary version 3". Nucleic Acids Research. 29 (1): 55–7. doi:10.1093/nar/29.1.55. PMC 29815. PMID 11125048.
- Dyson HJ, Sayre JR, Merutka G, Shin HC, Lerner RA, Wright PE (August 1992). "Folding of peptide fragments comprising the complete sequence of proteins. Models for initiation of protein folding. II. Plastocyanin". Journal of Molecular Biology. 226 (3): 819–35. doi:10.1016/0022-2836(92)90634-V. PMID 1507228.
- Fersht AR (February 1997). "Nucleation mechanisms in protein folding". Current Opinion in Structural Biology. 7 (1): 3–9. doi:10.1016/S0959-440X(97)80002-4. PMID 9032066.
- George DG, Hunt LT, Barker WC (1996). "PIR-International Protein Sequence Database". Methods in Enzymology. 266: 41–59. doi:10.1016/S0076-6879(96)66005-4. ISBN 978-0-12-182167-8. PMC 145575. PMID 8743676.
- Go M (May 1981). "Correlation of DNA exonic regions with protein structural units in haemoglobin". Nature. 291 (5810): 90–2. Bibcode:1981Natur.291...90G. doi:10.1038/291090a0. PMID 7231530. S2CID 4313732.
- Hadley C, Jones DT (September 1999). "A systematic comparison of protein structure classifications: SCOP, CATH and FSSP". Structure. 7 (9): 1099–112. doi:10.1016/S0969-2126(99)80177-4. PMID 10508779.
- Hayward S (September 1999). "Structural principles governing domain motions in proteins". Proteins. 36 (4): 425–35. doi:10.1002/(SICI)1097-0134(19990901)36:4<425::AID-PROT6>3.0.CO;2-S. PMID 10450084.
- Heringa J, Argos P (July 1991). "Side-chain clusters in protein structures and their role in protein folding". Journal of Molecular Biology. 220 (1): 151–71. doi:10.1016/0022-2836(91)90388-M. PMID 2067014.
- Honig B (October 1999). "Protein folding: from the levinthal paradox to structure prediction". Journal of Molecular Biology. 293 (2): 283–93. CiteSeerX 10.1.1.332.955. doi:10.1006/jmbi.1999.3006. PMID 10550209.
- Kim PS, Baldwin RL (1990). "Intermediates in the folding reactions of small proteins". Annual Review of Biochemistry. 59 (1): 631–60. doi:10.1146/annurev.bi.59.070190.003215. PMID 2197986.
- Murvai J, Vlahovicek K, Barta E, Cataletto B, Pongor S (January 2000). "The SBASE protein domain library, release 7.0: a collection of annotated protein sequence segments". Nucleic Acids Research. 28 (1): 260–2. doi:10.1093/nar/28.1.260. PMC 102474. PMID 10592241.
- Murzin AG, Brenner SE, Hubbard T, Chothia C (April 1995). "SCOP: a structural classification of proteins database for the investigation of sequences and structures" (PDF). Journal of Molecular Biology. 247 (4): 536–40. doi:10.1016/S0022-2836(05)80134-2. PMID 7723011. Archived from the original (PDF) on 26 April 2012.
- Janin J, Chothia C (1985). "Domains in proteins: definitions, location, and structural principles". Methods in Enzymology. 115: 420–30. doi:10.1016/0076-6879(85)15030-5. ISBN 978-0-12-182015-2. PMID 4079796.
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P (January 2000). "SMART: a web-based tool for the study of genetically mobile domains". Nucleic Acids Research. 28 (1): 231–4. doi:10.1093/nar/28.1.231. PMC 102444. PMID 10592234.
- Siddiqui AS, Dengler U, Barton GJ (February 2001). "3Dee: a database of protein structural domains". Bioinformatics. 17 (2): 200–1. doi:10.1093/bioinformatics/17.2.200. PMID 11238081.
- Srinivasarao GY, Yeh LS, Marzec CR, Orcutt BC, Barker WC, Pfeiffer F (January 1999). "Database of protein sequence alignments: PIR-ALN". Nucleic Acids Research. 27 (1): 284–5. doi:10.1093/nar/27.1.284. PMC 148157. PMID 9847202.
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, et al. (January 2001). "The COG database: new developments in phylogenetic classification of proteins from complete genomes". Nucleic Acids Research. 29 (1): 22–8. doi:10.1093/nar/29.1.22. PMC 29819. PMID 11125040.
- Taylor WR, Orengo CA (July 1989). "Protein structure alignment". Journal of Molecular Biology. 208 (1): 1–22. doi:10.1016/0022-2836(89)90084-3. PMID 2769748.
- Yang AS, Honig B (September 1995). "Free energy determinants of secondary structure formation: I. alpha-Helices". Journal of Molecular Biology. 252 (3): 351–65. doi:10.1006/jmbi.1995.0502. PMID 7563056.
- Yang AS, Honig B (September 1995). "Free energy determinants of secondary structure formation: II. Antiparallel beta-sheets". Journal of Molecular Biology. 252 (3): 366–76. doi:10.1006/jmbi.1995.0503. PMID 7563057.
- Gough J, Chothia C (January 2002). "SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments". Nucleic Acids Research. 30 (1): 268–72. doi:10.1093/nar/30.1.268. PMC 99153. PMID 11752312.
External links
Structural domain databases
Sequence domain databases
- InterPro
- Pfam at the Library of Congress Web Archives (archived 2011-05-06)
- PROSITE
- ProDom
- SMART
- NCBI Conserved Domain Database
- SUPERFAMILY Library of HMMs representing superfamilies and database of (superfamily and family) annotations for all completely sequenced organisms
Functional domain databases
- dcGO A comprehensive database of domain-centric ontologies on functions, phenotypes and diseases.