Sulfur cycle
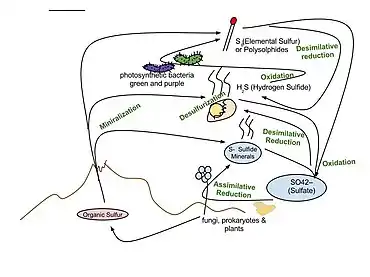
The sulfur cycle is a biogeochemical cycle in which the sulfur moves between rocks, waterways and living systems. It is important in geology as it affects many minerals and in life because sulfur is an essential element (CHNOPS), being a constituent of many proteins and cofactors, and sulfur compounds can be used as oxidants or reductants in microbial respiration.[1] The global sulfur cycle involves the transformations of sulfur species through different oxidation states, which play an important role in both geological and biological processes. Steps of the sulfur cycle are:
- Mineralization of organic sulfur into inorganic forms, such as hydrogen sulfide (H2S), elemental sulfur, as well as sulfide minerals.
- Oxidation of hydrogen sulfide, sulfide, and elemental sulfur (S) to sulfate (SO2−
4). - Reduction of sulfate to sulfide.
- Incorporation of sulfide into organic compounds (including metal-containing derivatives).
| Part of a series on |
| Biogeochemical cycles |
|---|
 |
|
These are often termed as follows:
- Assimilative sulfate reduction (see also sulfur assimilation) in which sulfate (SO2−
4) is reduced by plants, fungi and various prokaryotes. The oxidation states of sulfur are +6 in sulfate and –2 in R–SH. - Desulfurization in which organic molecules containing sulfur can be desulfurized, producing hydrogen sulfide gas (H2S, oxidation state = –2). An analogous process for organic nitrogen compounds is deamination.
- Oxidation of hydrogen sulfide produces elemental sulfur (S8), oxidation state = 0. This reaction occurs in the photosynthetic green and purple sulfur bacteria and some chemolithotrophs. Often the elemental sulfur is stored as polysulfides.
- Oxidation in elemental sulfur by sulfur oxidizers produces sulfate.
- Dissimilative sulfur reduction in which elemental sulfur can be reduced to hydrogen sulfide.
- Dissimilative sulfate reduction in which sulfate reducers generate hydrogen sulfide from sulfate.
Sulfur oxidation state
Sulfur has four main oxidation states in nature, which are −2, +2, +4, and +6. The common sulfur species of each oxidation state are listed as follows:
S2−: H2S, (CH3)2S, BaS
S0: native, or elemental, sulfur
S2+: SCl2
S4+: SO2, sulfite (SO2−
3)
S6+: SO2−
4 (H2SO4, CaSO4), SF6
Sulfur sources and sinks
Sulfur is found in oxidation states ranging from +6 in SO2−
4 to −2 in sulfides. Thus, elemental sulfur can either give or receive electrons depending on its environment. On the anoxic early Earth, most sulfur was present in minerals such as pyrite (FeS2). Over Earth history, the amount of mobile sulfur increased through volcanic activity as well as weathering of the crust in an oxygenated atmosphere.[1] Earth's main sulfur sink is the oceans SO2−
4, where it is the major oxidizing agent.[2]
| Food Types | Acidifying Emissions (g SO2eq per 100 g protein) |
|---|---|
| Beef | 343.6 |
| Cheese | 165.5 |
| Pork | 142.7 |
| Lamb and Mutton | 139.0 |
| Farmed Crustaceans | 133.1 |
| Poultry | 102.4 |
| Farmed Fish | 65.9 |
| Eggs | 53.7 |
| Groundnuts | 22.6 |
| Peas | 8.5 |
| Tofu | 6.7 |
When SO2−
4 is assimilated by organisms, it is reduced and converted to organic sulfur, which is an essential component of proteins. However, the biosphere does not act as a major sink for sulfur, instead the majority of sulfur is found in seawater or sedimentary rocks including: pyrite rich shales, evaporite rocks (anhydrite and baryte), and calcium and magnesium carbonates (i.e. carbonate-associated sulfate). The amount of sulfate in the oceans is controlled by three major processes:[4]
- input from rivers
- sulfate reduction and sulfide re-oxidation on continental shelves and slopes
- burial of anhydrite and pyrite in the oceanic crust.
The primary natural source of sulfur to the atmosphere is sea spray or windblown sulfur-rich dust,[5] neither of which is long lived in the atmosphere. In recent times, the large annual input of sulfur from the burning of coal and other fossil fuels has added a substantial amount SO2 which acts as an air pollutant. In the geologic past, igneous intrusions into coal measures have caused large scale burning of these measures, and consequential release of sulfur to the atmosphere. This has led to substantial disruption to the climate system, and is one of the proposed causes of the Permian–Triassic extinction event.
Dimethylsulfide [(CH3)2S or DMS] is produced by the decomposition of dimethylsulfoniopropionate (DMSP) from dying phytoplankton cells in the ocean's photic zone, and is the major biogenic gas emitted from the sea, where it is responsible for the distinctive “smell of the sea” along coastlines.[1] DMS is the largest natural source of sulfur gas, but still only has a residence time of about one day in the atmosphere and a majority of it is redeposited in the oceans rather than making it to land. However, it is a significant factor in the climate system, as it is involved in the formation of clouds.
Biologically and thermochemically driven sulfate reduction

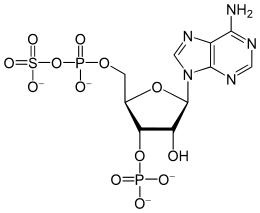
(key intermediate in the sulfur cycle)
Through the dissimilatory sulfate reduction pathway, sulfate can be reduced either bacterially (bacterial sulfate reduction) or inorganically (thermochemical sulfate reduction). This pathway involves the reduction of sulfate by organic compounds to produce hydrogen sulfide, which occurs in both processes.
The main products and reactants of bacterial sulfate reduction (BSR) and thermochemical sulfate reduction (TSR) are very similar. For both, various organic compounds and dissolved sulfate are the reactants, and the products or by-products are as follows: H2S, CO2, carbonates, elemental sulfur and metal sulfides.[6] However, the reactive organic compounds differ for BSR and TSR because of the mutually exclusive temperature regimes. Organic acids are the main organic reactants for BSR and branched/n-alkanes are the main organic reactants for TSR. The inorganic reaction products in BSR and TSR are H2S (HS−) and HCO−
3 (CO2).[7]
These processes occur because there are two very different thermal regimes in which sulfate is reduced, particularly in low-temperature and high-temperature environments.[6] BSR usually occurs at lower temperatures from 0−80 °C, while TSR happens at much higher temperatures around 100–140 °C.[7] Temperatures for TSR are not as well defined; the lowest confirmed temperature is 127 °C and the highest temperatures occur in settings around 160−180 °C.[7] These two different regimes appear because at higher temperatures most sulfate-reducing microbes can no longer metabolize due to the denaturation of proteins or deactivation of enzymes,[8] so TSR takes over. However, in hot sediments around hydrothermal vents BSR can happen at temperatures up to 110 °C.[9]
BSR and TSR occur at different depths. BSR takes place in low-temperature environments, which are shallower settings such as oil and gas fields. BSR can also take place in modern marine sedimentary environments such as stratified inland seas, continental shelves, organic-rich deltas, and hydrothermal sediments which have intense microbial sulfate reduction because of the high concentration of dissolved sulfate in the seawater.[10] Additionally, the high amounts of hydrogen sulfide found in oil and gas fields is thought to arise from the oxidation of petroleum hydrocarbons by sulfate.[11] Such reactions are known to occur by microbial processes but it is generally accepted that TSR is responsible for the bulk of these reactions, especially in deep or hot reservoirs.[12] Thus, TSR occurs in deep reservoirs where the temperatures are much higher. BSR is geologically instantaneous in most geologic settings, while TSR occurs at rates in the order of hundreds of thousands of years.[13][6] Although much slower than BSR, even TSR appears to be a geologically fairly fast process.
BSR in shallow environments and TSR in deep reservoirs are key processes in the oceanic sulfur cycle.[14][6] Approximately, 10% (of the total gas) of H2S is produced in BSR settings, whereas 90% of the H2S is produced in TSR settings.[7] If there is more than a few percent of H2S in any deep reservoir, then it is assumed that TSR has taken over. This is due to the fact that thermal cracking of hydrocarbons doesn't provide more than a couple percent of H2S. The amount of H2S is affected by several factors such as, the availability of organic reactants and sulfate and the presence/availability of base and transition metals.[15]
Sulfur-oxidizing bacteria in hydrothermal vents
Hydrothermal vents emit hydrogen sulfide that support the carbon fixation of chemolithotrophic bacteria that oxidize hydrogen sulfide with oxygen to produce elemental sulfur or sulfate.[16] The chemical reactions are as follows:
- CO2 + 4 H2S + O2 → CH2O + 4 S0 + 3 H2O
- CO2 + H2S + O2 + H2O → CH2O + SO2–
4 + 2 H+
In modern oceans, Thiomicrospira, Halothiobacillus, and Beggiatoa are primary sulfur oxidizing bacteria,[16] and form chemosynthetic symbioses with animal hosts.[17] The host provides metabolic substrates (e.g., CO2, O2, H2O) to the symbiont while the symbiont generates organic carbon for sustaining the metabolic activities of the host. The produced sulfate usually combines with the leached calcium ions to form gypsum, which can form widespread deposits on near mid-ocean spreading centers.[18]
δ34S
Although there are 25 known isotopes of sulfur, only four are stable and of geochemical importance. Of those four, two (32S, light and 34S, heavy) comprise (99.22%) of sulfur on Earth. The vast majority (95.02%) of sulfur occurs as 32S with only 4.21% in 34S. The ratio of these two isotopes is fixed in the Solar System and has been since its formation. The bulk Earth sulfur isotopic ratio is thought to be the same as the ratio of 22.22 measured from the Canyon Diablo troilite (CDT), a meteorite.[19] That ratio is accepted as the international standard and is therefore set at δ = 0.00. Deviation from 0.00 is expressed as the δ34S which is a ratio in per mill (‰). Positive values correlate to increased levels of 34S, whereas negative values correlate with greater 32S in a sample.
Formation of sulfur minerals through non-biogenic processes does not substantially differentiate between the light and heavy isotopes, therefore sulfur isotope ratios in gypsum or barite should be the same as the overall isotope ratio in the water column at their time of precipitation. Sulfate reduction through biologic activity strongly differentiates between the two isotopes because of the more rapid enzymic reaction with 32S.[19] Sulfate metabolism results in an isotopic depletion of −18‰, and repeated cycles of oxidation and reduction can result in values up to −50‰. Average present day seawater values of δ34S are on the order of +21‰.
Throughout geologic history the sulfur cycle and the isotopic ratios have coevolved with the biosphere becoming overall more negative with the increases in biologically driven sulfate reduction, but also show substantial positive excursion. In general positive excursions in the sulfur isotopes mean that there is an excess of pyrite deposition rather than oxidation of sulfide minerals exposed on land.[19]
Marine sulfur cycle
The sulfur cycle in marine environments has been well-studied via the tool of sulfur isotope systematics expressed as δ34S. The modern global oceans have sulfur storage of 1.3×1018 kg,[20] mainly occurring as sulfate with the δ34S value of +21‰.[21] The overall input flux is 1.0×1011 kg/a with the sulfur isotope composition of ~3‰.[21] Riverine sulfate derived from the terrestrial weathering of sulfide minerals (δ34S = +6‰) is the primary input of sulfur to the oceans. Other sources are metamorphic and volcanic degassing and hydrothermal activity (δ34S = 0‰), which release reduced sulfur species (such as H2S and S0). There are two major outputs of sulfur from the oceans. The first sink is the burial of sulfate either as marine evaporites (such as gypsum) or carbonate-associated sulfate (CAS), which accounts for 6×1010 kg/a (δ34S = +21‰). The second sulfur sink is pyrite burial in shelf sediments or deep seafloor sediments (4×1010 kg/a; δ34S = −20‰).[22] The total marine sulfur output flux is 1.0×1011 kg/a which matches the input fluxes, implying the modern marine sulfur budget is at steady state.[21] The residence time of sulfur in modern global oceans is 13,000,000 years.[23]
Evolution of the sulfur cycle
The isotopic composition of sedimentary sulfides provides primary information on the evolution of the sulfur cycle.
The total inventory of sulfur compounds on the surface of the Earth (nearly 1019 kg of sulfur) represents the total outgassing of sulfur through geologic time.[24][19] Rocks analyzed for sulfur content are generally organic-rich shales meaning they are likely controlled by biogenic sulfur reduction. Average seawater curves are generated from evaporites deposited throughout geologic time because again, since they do not discriminate between the heavy and light sulfur isotopes, they should mimic the ocean composition at the time of deposition.
4.6 billion years ago (Ga) the Earth formed and had a theoretical δ34S value of 0. Since there was no biologic activity on early Earth there would be no isotopic fractionation.[21] All sulfur in the atmosphere would be released during volcanic eruptions. When the oceans condensed on Earth, the atmosphere was essentially swept clean of sulfur gases, owing to their high solubility in water. Throughout the majority of the Archean (4.6–2.5 Ga) most systems appeared to be sulfate-limited. Some small Archean evaporite deposits require that at least locally elevated concentrations (possibly due to local volcanic activity) of sulfate existed in order for them to be supersaturated and precipitate out of solution.[25]
3.8–3.6 Ga marks the beginning of the exposed geologic record because this is the age of the oldest rocks on Earth. Metasedimentary rocks from this time still have an isotopic value of 0 because the biosphere was not developed enough (possibly at all) to fractionate sulfur.[26]
3.5 Ga anoxyogenic photosynthesis is established and provides a weak source of sulfate to the global ocean with sulfate concentrations incredibly low the δ34S is still basically 0.[25] Shortly after, at 3.4 Ga the first evidence for minimal fractionation in evaporitic sulfate in association with magmatically derived sulfides can be seen in the rock record. This fractionation shows possible evidence for anoxygenic phototrophic bacteria.
2.8 Ga marks the first evidence for oxygen production through photosynthesis. This is important because there cannot be sulfur oxidation without oxygen in the atmosphere. This exemplifies the coevolution of the oxygen and sulfur cycles as well as the biosphere.
2.7–2.5 Ga is the age of the oldest sedimentary rocks to have a depleted δ 34S which provide the first compelling evidence for sulfate reduction.[25]
2.3 Ga sulfate increases to more than 1 mM; this increase in sulfate is coincident with the "Great Oxygenation Event", when redox conditions on Earth's surface are thought by most workers to have shifted fundamentally from reducing to oxidizing.[27] This shift would have led to an incredible increase in sulfate weathering which would have led to an increase in sulfate in the oceans. The large isotopic fractionations that would likely be associated with bacteria reduction are produced for the first time. Although there was a distinct rise in seawater sulfate at this time it was likely still only less than 5–15% of present-day levels.[27]
At 1.8 Ga, Banded iron formations (BIF) are common sedimentary rocks throughout the Archean and Paleoproterozoic; their disappearance marks a distinct shift in the chemistry of ocean water. BIFs have alternating layers of iron oxides and chert. BIFs only form if the water is allowed to supersaturate in dissolved iron (Fe2+) meaning there cannot be free oxygen or sulfur in the water column because it would form Fe3+ (rust) or pyrite and precipitate out of solution. Following this supersaturation, the water must become oxygenated in order for the ferric rich bands to precipitate it must still be sulfur poor otherwise pyrite would form instead of Fe3+. It has been hypothesized that BIFs formed during the initial evolution of photosynthetic organisms that had phases of population growth, causing over production of oxygen. Due to this over production they would poison themselves causing a mass die off, which would cut off the source of oxygen and produce a large amount of CO2 through the decomposition of their bodies, allowing for another bacterial bloom. After 1.8 Ga sulfate concentrations were sufficient to increase rates of sulfate reduction to greater than the delivery flux of iron to the oceans.[25]
Along with the disappearance of BIF, the end of the Paleoproterozoic also marks the first large scale sedimentary exhalative deposits showing a link between mineralization and a likely increase in the amount of sulfate in sea water. In the Paleoproterozoic the sulfate in seawater had increased to an amount greater than in the Archean, but was still lower than present day values.[27] The sulfate levels in the Proterozoic also act as proxies for atmospheric oxygen because sulfate is produced mostly through weathering of the continents in the presence of oxygen. The low levels in the Proterozoic simply imply that levels of atmospheric oxygen fell between the abundances of the Phanerozoic and the deficiencies of the Archean.
750 million years ago (Ma) there is a renewed deposition of BIF which marks a significant change in ocean chemistry. This was likely due to snowball Earth episodes where the entire globe including the oceans was covered in a layer of ice cutting off oxygenation.[28] In the late Neoproterozoic high carbon burial rates increased the atmospheric oxygen level to >10% of its present-day value. In the Latest Neoproterozoic another major oxidizing event occurred on Earth's surface that resulted in an oxic deep ocean and possibly allowed for the appearance of multicellular life.[27]
During the last 600 million years, seawater SO4 has generally varied between +10‰ and +30‰ in δ34S, with an average value close to that of today. Notably changes in seawater δ34S occurred during extinction and climatic events during this time.[29][30][31][32][33][34][35]
Over a shorter time scale (ten million years) changes in the sulfur cycle are easier to observe and can be even better constrained with oxygen isotopes. Oxygen is continually incorporated into the sulfur cycle through sulfate oxidation and then released when that sulfate is reduced once again.[4] Since different sulfate sources within the ocean have distinct oxygen isotopic values it may be possible to use oxygen to trace the sulfur cycle. Biological sulfate reduction preferentially selects lighter oxygen isotopes for the same reason that lighter sulfur isotopes are preferred. By studying oxygen isotopes in ocean sediments over the last 10 million years[36] were able to better constrain the sulfur concentrations in sea water through that same time. They found that the sea level changes due to Pliocene and Pleistocene glacial cycles changed the area of continental shelves which then disrupted the sulfur processing, lowering the concentration of sulfate in the sea water. This was a drastic change as compared to preglacial times before 2 million years ago.
The Great Oxidation Event and sulfur isotope mass-independent fractionation
The Great Oxygenation Event (GOE) is characterized by the disappearance of sulfur isotope mass-independent fractionation (MIF) in the sedimentary records at around 2.45 billion years ago (Ga).[37] The MIF of sulfur isotope (Δ33S) is defined by the deviation of measured δ33S value from the δ33S value inferred from the measured δ34S value according to the mass dependent fractionation law. The Great Oxidation Event represented a massive transition of global sulfur cycles. Before the Great Oxidation Event, the sulfur cycle was heavily influenced by the ultraviolet (UV) radiation and the associated photochemical reactions, which induced the sulfur isotope mass-independent fractionation (Δ33S ≠ 0). The preservation of sulfur isotope mass-independent fractionation signals requires the atmospheric O2 lower than 10−5 of present atmospheric level (PAL).[24] The disappearance of sulfur isotope mass-independent fractionation at ~2.45 Ga indicates that atmospheric pO2 exceeded 10−5 present atmospheric level after the Great Oxygenation Event.[37] Oxygen played an essential role in the global sulfur cycles after the Great Oxygenation Event, such as oxidative weathering of sulfides.[38] The burial of pyrite in sediments in turn contributes to the accumulation of free O2 in Earth's surface environment.[39]
Economic importance
Sulfur is intimately involved in production of fossil fuels and a majority of metal deposits because of its ability to act as an oxidizing or reducing agent. The vast majority of the major mineral deposits on Earth contain a substantial amount of sulfur including, but not limited to: sedimentary exhalative deposits (SEDEX), Carbonate-hosted lead-zinc ore deposits (Mississippi Valley-Type MVT) and porphyry copper deposits. Iron sulfides, galena and sphalerite will form as by-products of hydrogen sulfide generation, as long as the respective transition or base metals are present or transported to a sulfate reduction site.[7] If the system runs out of reactive hydrocarbons economically viable elemental sulfur deposits may form. Sulfur also acts as a reducing agent in many natural gas reservoirs and generally ore forming fluids have a close relationship with ancient hydrocarbon seeps or vents.[27]
Important sources of sulfur in ore deposits are generally deep-seated, but they can also come from local country rocks, sea water, or marine evaporites. The presence or absence of sulfur is one of the limiting factors on both the concentration of precious metals and its precipitation from solution. pH, temperature and especially redox states determine whether sulfides will precipitate. Most sulfide brines will remain in concentration until they reach reducing conditions, a higher pH or lower temperatures.
Ore fluids are generally linked to metal rich waters that have been heated within a sedimentary basin under the elevated thermal conditions typically in extensional tectonic settings. The redox conditions of the basin lithologies exert an important control on the redox state of the metal-transporting fluids and deposits can form from both oxidizing and reducing fluids.[27] Metal-rich ore fluids tend to be by necessity comparatively sulfide deficient, so a substantial portion of the sulfide must be supplied from another source at the site of mineralization. Bacterial reduction of seawater sulfate or a euxinic (anoxic and H2S-containing) water column is a necessary source of that sulfide. When present, the δ34S values of barite are generally consistent with a seawater sulfate source, suggesting baryte formation by reaction between hydrothermal barium and sulfate in ambient seawater.[27]
Once fossil fuels or precious metals are discovered and either burned or milled, the sulfur become a waste product which must be dealt with properly or it can become a pollutant. There has been a great increase in the amount of sulfur in our present day atmosphere because of the burning of fossil fuels. Sulfur acts as a pollutant and an economic resource at the same time.
Human impact
Human activities have a major effect on the global sulfur cycle. The burning of coal, natural gas, and other fossil fuels has greatly increased the amount of sulfur in the atmosphere and ocean and depleted the sedimentary rock sink. Without human impact sulfur would stay tied up in rocks for millions of years until it was uplifted through tectonic events and then released through erosion and weathering processes. Instead it is being drilled, pumped and burned at a steadily increasing rate. Over the most polluted areas there has been a 30-fold increase in sulfate deposition.[40]
Although the sulfur curve shows shifts between net sulfur oxidation and net sulfur reduction in the geologic past, the magnitude of the current human impact is probably unprecedented in the geologic record. Human activities greatly increase the flux of sulfur to the atmosphere, some of which is transported globally. Humans are mining coal and extracting petroleum from the Earth's crust at a rate that mobilizes 150 x 1012 gS/yr, which is more than double the rate of 100 years ago.[41] The result of human impact on these processes is to increase the pool of oxidized sulfur (SO4) in the global cycle, at the expense of the storage of reduced sulfur in the Earth's crust. Therefore, human activities do not cause a major change in the global pools of sulfur, but they do produce massive changes in the annual flux of sulfur through the atmosphere.[19]
When SO2 is emitted as an air pollutant, it forms sulfuric acid through reactions with water in the atmosphere. Once the acid is completely dissociated in water the pH can drop to 4.3 or lower causing damage to both man-made and natural systems. According to the EPA, acid rain is a broad term referring to a mixture of wet and dry deposition (deposited material) from the atmosphere containing higher than normal amounts of nitric and sulfuric acids. Distilled water (water without any dissolved constituents), which contains no carbon dioxide, has a neutral pH of 7. Rain naturally has a slightly acidic pH of 5.6, because carbon dioxide and water in the air react together to form carbonic acid, a very weak acid. Around Washington, D.C., however, the average rain pH is between 4.2 and 4.4. Since pH is on a log scale dropping by 1 (the difference between normal rain water and acid rain) has a dramatic effect on the strength of the acid. In the United States, roughly two thirds of all SO2 and one fourth of all NO3 come from electric power generation that relies on burning fossil fuels, like coal.
As it is an important nutrient for plants, sulfur is increasingly used as a component of fertilizers. Recently sulfur deficiency has become widespread in many countries in Europe.[42][43][44] Because of actions taken to limit acid rains atmospheric inputs of sulfur continue to decrease, As a result, the deficit in the sulfur input is likely to increase unless sulfur fertilizers are used.[45][46]
See also
References
- Madigan MT, Martino JM (2006). Brock Biology of Microorganisms (11th ed.). Pearson. p. 136. ISBN 978-0-13-196893-6.
- Bickle MJ, Alt JC, Teagle DA (1994). "Sulfur transport and sulphur isotope fractionations in ocean floor hydrothermal systems". Mineralogical Magazine. 58A (1): 88–89. Bibcode:1994MinM...58...88B. doi:10.1180/minmag.1994.58A.1.49.
- Poore J, Nemecek T (June 2018). "Reducing food's environmental impacts through producers and consumers". Science. 360 (6392): 987–992. Bibcode:2018Sci...360..987P. doi:10.1126/science.aaq0216. PMID 29853680.
- Turchyn, Alexandra V. (2005). Oxygen isotopes in marine sulfate and the sulfur cycle over the last 140 million years (PhD). Harvard University. 3174055.
- Reheis MC, Kihl R (May 1995). "Dust deposition in southern Nevada and California, 1984–1989: Relations to climate, source area, and source lithology". Journal of Geophysical Research: Atmospheres. 100 (D5): 8893–8918. Bibcode:1995JGR...100.8893R. doi:10.1029/94JD03245.
- Machel HG, Krouse HR, Sassen R (1995). "Products and distinguishing criteria of bacterial and thermochemical sulfate reduction". Applied Geochemistry. 10 (4): 373–389. Bibcode:1995ApGC...10..373M. doi:10.1016/0883-2927(95)00008-8.
- Machel HG (2001). "Bacterial and thermochemical sulfate reduction in diagenetic settings — old and new insights". Sedimentary Geology. 140 (1–2): 143–175. Bibcode:2001SedG..140..143M. doi:10.1016/S0037-0738(00)00176-7.
- Barton L (1995). Sulfate-reducing bacteria. New York: Plenum Press. ISBN 0-306-44857-2. OCLC 32311676.
- Jørgensen BB, Isaksen MF, Jannasch HW (December 1992). "Bacterial Sulfate Reduction Above 100{degrees}C in Deep-Sea Hydrothermal Vent Sediments". Science. 258 (5089): 1756–1757. doi:10.1126/science.258.5089.1756. PMID 17831655. S2CID 129371120.
- Aharon P, Fu B (2000). "Microbial sulfate reduction rates and sulfur and oxygen isotope fractionations at oil and gas seeps in deepwater Gulf of Mexico". Geochimica et Cosmochimica Acta. 64 (2): 233–246. Bibcode:2000GeCoA..64..233A. doi:10.1016/S0016-7037(99)00292-6.
- Goldstein TP, Aizenshtat Z (1994). "Thermochemical sulfate reduction a review". Journal of Thermal Analysis. 42 (1): 241–290. doi:10.1007/BF02547004. ISSN 0368-4466. S2CID 95526523.
- Krouse HR, Viau CA, Eliuk LS, Ueda A, Halas S (1988). "Chemical and isotopic evidence of thermochemical sulphate reduction by light hydrocarbon gases in deep carbonate reservoirs". Nature. 333 (6172): 415–419. Bibcode:1988Natur.333..415K. doi:10.1038/333415a0. ISSN 0028-0836. S2CID 4354648.
- Muyzer G, Stams AJ (June 2008). "The ecology and biotechnology of sulphate-reducing bacteria". Nature Reviews. Microbiology. 6 (6): 441–454. doi:10.1038/nrmicro1892. PMID 18461075. S2CID 22775967.
- Jørgensen BB (1982). "Mineralization of organic matter in the sea bed—the role of sulphate reduction". Nature. 296 (5858): 643–645. Bibcode:1982Natur.296..643J. doi:10.1038/296643a0. ISSN 0028-0836. S2CID 4308770.
- Holmer M, Storkholm P (2001). "Sulphate reduction and sulphur cycling in lake sediments: a review". Freshwater Biology. 46 (4): 431–451. doi:10.1046/j.1365-2427.2001.00687.x. ISSN 0046-5070.
- Sievert SM, Hügler M, Taylor CD, Wirsen CO (2008). Dahl C, Friedrich CG (eds.). "Sulfur Oxidation at Deep-Sea Hydrothermal Vents". Microbial Sulfur Metabolism. Springer Berlin Heidelberg: 238–258. doi:10.1007/978-3-540-72682-1_19. ISBN 978-3-540-72679-1.
- Klotz MG, Bryant DA, Hanson TE (2011). "The microbial sulfur cycle". Frontiers in Microbiology. 2: 241. doi:10.3389/fmicb.2011.00241. PMC 3228992. PMID 22144979.
- Pedersen RB, Rapp HT, Thorseth IH, Lilley MD, Barriga FJ, Baumberger T, et al. (November 2010). "Discovery of a black smoker vent field and vent fauna at the Arctic Mid-Ocean Ridge". Nature Communications. 1 (8): 126. Bibcode:2010NatCo...1..126P. doi:10.1038/ncomms1124. PMC 3060606. PMID 21119639.
- Schlesinger WH (1997). Biogeochemistry an analysis of global change (2nd ed.). San Diego, California: Academic Press. ISBN 9780126251555.
- Brimblecombe P (2014). "The global sulfur cycle". Treatise on Geochemistry. Vol. 10. Amsterdam: Elsevier. pp. 559–591. doi:10.1016/B978-0-08-095975-7.00814-7. ISBN 9780080983004.
- Fike DA, Bradley AS, Rose CV (2015). "Rethinking the Ancient Sulfur Cycle". Annual Review of Earth and Planetary Sciences. 43 (1): 593–622. Bibcode:2015AREPS..43..593F. doi:10.1146/annurev-earth-060313-054802. S2CID 140644882.
- Canfield DE (2004). "The evolution of the Earth surface sulfur reservoir". American Journal of Science. 304 (10): 839–861. Bibcode:2004AmJS..304..839C. doi:10.2475/ajs.304.10.839.
- Kah LC, Lyons TW, Frank TD (October 2004). "Low marine sulphate and protracted oxygenation of the Proterozoic biosphere". Nature. 431 (7010): 834–838. Bibcode:2004Natur.431..834K. doi:10.1038/nature02974. PMID 15483609. S2CID 4404486.
- Johnston DT (2011). "Multiple sulfur isotopes and the evolution of Earth's surface sulfur cycle". Earth-Science Reviews. 106 (1–2): 161–183. Bibcode:2011ESRv..106..161J. doi:10.1016/j.earscirev.2011.02.003.
- Canfield DE, Raiswell R (1999). "The evolution of the sulfur cycle". American Journal of Science. 299 (7–9): 697–723. Bibcode:1999AmJS..299..697C. doi:10.2475/ajs.299.7-9.697.
- Schidlowski M, Hayes JM, Kaplan IR (1983). "Isotopic inferences of ancient biochemistries-Carbon, sulfur, hydrogen, and nitrogen.". In Schopf JW (ed.). Earth's Earliest Biosphere. Princeton, New Jersey: Princeton University Press.
- Lyons TW, Gellatly AM, McGoldrick PJ, Kah LC (2006). "Proterozoic sedimentary exhalative (SEDEX) deposits and links to evolving global ocean chemistry". In Kesler SE, Ohmoto H (eds.). Evolution of Early Earth's Atmosphere, Hydrosphere, and Biosphere—Constraints from Ore Deposits. Geological Society of America Memoir. Vol. 198. pp. 169–184. ISBN 978-0-8137-1198-0.
- Hoffman PF, Kaufman AJ, Halverson GP, Schrag DP (August 1998). "A neoproterozoic snowball earth". Science. 281 (5381): 1342–1346. Bibcode:1998Sci...281.1342H. doi:10.1126/science.281.5381.1342. PMID 9721097. S2CID 13046760.
- Gill BC, Lyons TW, Young SA, Kump LR, Knoll AH, Saltzman MR (January 2011). "Geochemical evidence for widespread euxinia in the later Cambrian ocean". Nature. 469 (7328): 80–83. Bibcode:2011Natur.469...80G. doi:10.1038/nature09700. PMID 21209662. S2CID 4319979.
- John EH, Wignall PB, Newton RJ, Bottrell SH (August 2010). "δ34SCAS and δ18OCAS records during the Frasnian–Famennian (Late Devonian) transition and their bearing on mass extinction models". Chemical Geology. 275 (3–4): 221–234. Bibcode:2010ChGeo.275..221J. doi:10.1016/j.chemgeo.2010.05.012.
- Newton RJ, Pevitt EL, Wignall PB, Bottrell SH (February 2004). "Large shifts in the isotopic composition of seawater sulphate across the Permo–Triassic boundary in northern Italy". Earth and Planetary Science Letters. 218 (3–4): 331–345. Bibcode:2004E&PSL.218..331N. doi:10.1016/S0012-821X(03)00676-9.
- Gill BC, Lyons TW, Jenkyns HC (December 2011). "A global perturbation to the sulfur cycle during the Toarcian Oceanic Anoxic Event". Earth and Planetary Science Letters. 312 (3–4): 484–496. Bibcode:2011E&PSL.312..484G. doi:10.1016/j.epsl.2011.10.030.
- Paytan, A. (1998-11-20). "Sulfur Isotopic Composition of Cenozoic Seawater Sulfate". Science. 282 (5393): 1459–1462. doi:10.1126/science.282.5393.1459. PMID 9822370.
- Paytan, A. (2004-06-11). "Seawater Sulfur Isotope Fluctuations in the Cretaceous". Science. 304 (5677): 1663–1665. Bibcode:2004Sci...304.1663P. doi:10.1126/science.1095258. ISSN 0036-8075. PMID 15192227. S2CID 10539452.
- Owens JD, Gill BC, Jenkyns HC, Bates SM, Severmann S, Kuypers MM, et al. (November 2013). "Sulfur isotopes track the global extent and dynamics of euxinia during Cretaceous Oceanic Anoxic Event 2". Proceedings of the National Academy of Sciences of the United States of America. 110 (46): 18407–18412. Bibcode:2013PNAS..11018407O. doi:10.1073/pnas.1305304110. PMC 3831968. PMID 24170863.
- Tychyn et al. (2004) incomplete ref
- Farquhar J, Bao H, Thiemens M (August 2000). "Atmospheric influence of Earth's earliest sulfur cycle". Science. 289 (5480): 756–759. Bibcode:2000Sci...289..756F. doi:10.1126/science.289.5480.756. PMID 10926533. S2CID 12287304.
- Konhauser KO, Lalonde SV, Planavsky NJ, Pecoits E, Lyons TW, Mojzsis SJ, et al. (October 2011). "Aerobic bacterial pyrite oxidation and acid rock drainage during the Great Oxidation Event". Nature. 478 (7369): 369–373. Bibcode:2011Natur.478..369K. doi:10.1038/nature10511. PMID 22012395. S2CID 205226545.
- Berner RA, Raiswell R (1983). "Burial of organic carbon and pyrite sulfur in sediments over phanerozoic time: a new theory". Geochimica et Cosmochimica Acta. 47 (5): 855–862. Bibcode:1983GeCoA..47..855B. doi:10.1016/0016-7037(83)90151-5.
- Pham M, Müller JF, Brasseur GP, Granier C, Mégie G (May 1996). "A 3D model study of the global sulphur cycle: Contributions of anthropogenic and biogenic sources". Atmospheric Environment. 30 (10–11): 1815–1822. Bibcode:1996AtmEn..30.1815P. doi:10.1016/1352-2310(95)00390-8.
- Brimblecombe P, Hammer C, Rodhe H, Ryaboshapko A, Boutron CF (1989). "Human Influences on the sulphur cycle.". In Brimblecombe P, Lein AY (eds.). Evolution of the Global Biogeochemical Sulphur Cycle. New York: Wiley. pp. 77–121. ISBN 978-0-471-92251-3.
- Zhao F, Hawkesford M, McGrath SP (1999). "Sulphur Assimilation and Effects on Yield and Quality of Wheat". Journal of Cereal Science. 30 (1): 1–17. doi:10.1006/jcrs.1998.0241.
- Blake-Kalff MM (2000). "Diagnosing sulfur deficiency in field-grown oilseed rape (Brassica napus L.) and wheat (Triticum aestivum L.)". Plant and Soil. 225 (1−2): 95–107. doi:10.1023/A:1026503812267. S2CID 44208638.
- Ceccotti SP (1996). "Plant nutrient sulphur—a review of nutrient balance, environmental impact and fertilizers". Fertilizer Research. 43 (1–3): 117–125. doi:10.1007/BF00747690. S2CID 42207099.
- Glossary, United States: NASA Earth Observatory, acid rain, archived from the original on December 13, 2011, retrieved February 15, 2013
- Sulfur as a fertilizer. Sulphurinstitute.org. Retrieved on 16 August 2012.