Adición oxidante
La adición oxidante es un tipo de reacción química, relacionada con los procesos en la química organometálica y en la que aumenta tanto el estado de oxidación como el número de coordinación de un centro metálico.[1][2][3] El proceso de adición oxidante se emplea a menudo en ciclos catalíticos, junto con su reacción inversa, la eliminación reductora.[4]
En los metales de transición
Para los metales de transición, la reacción de oxidación origina la disminución de la configuración electrónica del orbital dn para pasar a un estado con menos electrones, habitualmente con 2 electrones menos. La adición oxidante se favorece con los metales que son básicos o fácilmente oxidables. Metales con un relativamente bajo estado de oxidación cumplen con este requisito, pero metales, incluso de alto estado de oxidación sufren la adición oxidante, como se ilustra en la oxidación de Pt (II) con el cloro:
- [PtCl4]2- + Cl2 → [PtCl6]2-
En química organometálica clásica, el estado de oxidación formal del metal y el número de electrones del complejo se incrementan en dos unidades[5] Los cambios de un solo electrón también son posibles y, de hecho, algunas de las reacciones de adición oxidante proceden a través de una serie de cambios monoelectrónicos. Aunque las adiciones oxidantes pueden transcurrir con la inserción de un metal en muchos sustratos diferentes, se observan con mayor frecuencia con H-H, H-X, y los enlaces C-X porque estos sustratos son relevantes para las aplicaciones comerciales.
La adición oxidante requiere que el complejo metálico tenga un punto de coordinación vacante. Por esta razón, las adiciones oxidantes son comunes para los complejos con índice de coordinación cuatro y cinco.
La eliminación reductiva es la reacción inversa de la adición oxidante.[6] Se favorece la eliminación reductora cuando el recién formado enlace X-Y es fuerte. Para que tenga lugar la eliminación reductiva, los dos grupos (X e Y) debe ser mutuamente adyacentes en la esfera de coordinación del metal. La eliminación reductiva es la etapa clave en la liberación del producto en varias reacciones que forman enlaces C-C y C-H.[4]
Mecanismos de adición oxidante
Las adiciones oxidantes transcurren a través de muchos mecanismos diferentes que dependen del centro metálico y de los sustratos.
Mecanismo concertado
Las adiciones oxidantes de sustratos no polares, como el hidrógeno y los hidrocarburos, parecen proceder a través de mecanismos concertados. Tales sustratos no poseen enlaces pi, en consecuencia, se formaría un complejo σ de tres centros, seguido por la división del enlace intramolecular del ligando para formar el complejo oxidado. Los ligandos resultante será mutuamente cis,[1] aunque puede ocurrir una isomerización posterior.

Este mecanismo se aplica a la adición de moléculas diatómicas homonucleares, tales como H2. Muchas reacciones de activación C-H también siguen un mecanismo concertado a través de la formación de un complejo con enlace agóstico M-(C-H)[7][1]
Un ejemplo representativo es la reacción de hidrógeno con el complejo de Vaska, trans-IrCl(CO)[P(C6H5)3]2. En esta transformación, el iridio cambia su estado de oxidación formal de +I a +III. El producto está enlazado formalmente a tres aniones: un ligando cloruro y dos hidruros. Como se muestra a continuación, el complejo metálico inicial tiene 16 electrones de valencia y un número de coordinación de cuatro, mientras que el producto es un complejo con índice de coordenación seis y 18 electrones.
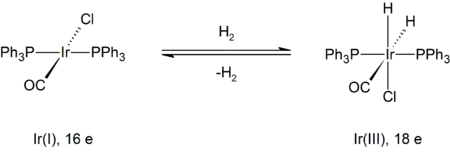
La formación de un intermedio di-hidrógeno con geometría bipiramidal trigonal está seguida por escisión del enlace H-H, debido a la retrodonación de electrones al orbital σ* del H-H.[8]
Este sistema también se encuentra en equilibrio químico con la reacción inversa continuando con la eliminación de gas hidrógeno con una reducción simultánea del centro metálico.
Ya que la división del enlace H-H requiere retrodonación de electrones al orbital σ* del H-H, los metales ricos en electrones favorecen esta reacción.[8] El mecanismo concertado produce un dihidruro cis, mientras la estereoquímica de otros mecanismos de adición oxidante no suelen producir aductos cis.
Mecanismo tipo SN2
Algunas adiciones oxidantes transcurren de manera análoga a las bien conocidas reacciones de sustitución nucleófila bimolecular en química orgánica. El ataque nucleofílico del centro metálico al átomo menos electronegativo del sustrato lleva a la escisión del enlace R-X, para formar la especie [M-R]+. Este paso es seguido por una rápida coordinación del anión al centro metálico catiónico. Por ejemplo, la reacción de un complejo cuadrado plano con yoduro de metilo:

Este mecanismo se supone a menudo en la adición de sustratos polares y electrófilos, tales como haluros de alquilo y halógenos.[1]
Mecanismo iónico
El mecanismo iónico de adición oxidante es similar al tipo SN2 ya que se trata de la adición gradual de dos fragmentos distintos de ligando. La diferencia clave es que los mecanismos iónicos incluyen sustratos que se disocian en solución antes de cualquier interacción con el centro metálico (por ejemplo, las adiciones de HCl en solución acuosa).[1]
Mecanismo radicalario
Además de las reacciones de tipo SN2, los halogenuros de alquilo y otros sustratos similares pueden agregarse a un centro metálico a través de un mecanismo radicalario. Sin embargo, ha habido controversia sobre la validez de los experimentos utilizados en la detección de intermedios radicales.[1] Sin embargo, son conocidas las reacciones que son generalmente aceptadas como continuación de un mecanismo mediante radicales. Un ejemplo fue propuesto por Lednor y colaboradores.[9]
- Iniciación
- [(CH3)2C(CN)N]2 → 2 (CH3)2(CN)C• + N2
- (CH3)2(CN)C• + PhBr → (CH3)2(CN)CBr + Ph•
- Propagación
- Ph• + [Pt(PPh3)2] → [Pt(PPh3)2Ph]•
- [Pt(PPh3)2Ph]• + PhBr → [Pt(PPh3)2PhBr] + Ph•
Aplicaciones
La adición oxidante y la eliminación reductiva participan en muchos procesos catalíticos, tanto en catálisis homogénea (es decir, en disolución) como en catálisis heterogénea, como el proceso Monsanto y la hidrogenación de alquenos usando el catalizador de Wilkinson.
Referencias
- Crabtree, Robert (2005). The Organometallic Chemistry of the Transition Metals. Wiley-Interscience. pp. 159–180. ISBN 0-471-66256-9.
- Inorganic Chemistry (3rd Edition) by Gary L. Miessler, Donald A. Tarr
- Inorganic Chemistry by D. F. Shriver, P. W. Atkins
- Hartwig, J. F. (2010). Organotransition Metal Chemistry, from Bonding to Catalysis. New York: University Science Books. ISBN 189138953X.
- Unión Internacional de Química Pura y Aplicada. «oxidative addition». Compendium of Chemical Terminology. Versión en línea (en inglés).
- Unión Internacional de Química Pura y Aplicada. «reductive elimination». Compendium of Chemical Terminology. Versión en línea (en inglés).
- El enlace M-H-C agóstico. Química Organometálica. Didier Astruc, Frederic Astruc. Editorial Reverté, 2003. ISBN 84-291-7007-3. Pág. 181
- Johnson, Curtis; Richard Eisenberg (1985). «Stereoselective Oxidative Addition of Hydrogen to Iridium(I) Complexes. Kinetic Control Based on Ligand Electronic Effects». Journal of the American Chemical Society 107 (11): 3148-3160. doi:10.1021/ja00297a021.
- Hall, Thomas L.; Michael F. Lappert, and Peter W. Lednor (1980). «Mechanistic studies of some oxidative-addition reactions: free-radical pathways in the Pt0-RX, Pt0-PhBr, and PtII-R'SO2X Reactions (R = alkyl, R' = aryl, X = halide) and in the related rhodium(I) or iridium(I) Systems». Journal of the Chemical Society, Dalton Transactions (8): 1448-1456. doi:10.1039/DT9800001448.
Lecturas adicionales
- Ananikov, Valentine P.; Musaev, Djamaladdin G.; Morokuma, Keiji (2005). «Theoretical Insight into the C−C Coupling Reactions of the Vinyl, Phenyl, Ethynyl, and Methyl Complexes of Palladium and Platinum». Organometallics 24 (4): 715. doi:10.1021/om0490841.
Enlaces externos
- R. Toreki. «Oxidative Addition». The Organometallic HyperTextBook. Interactive Learning Paradigms Inc.
- R. Toreki. «Reductive Elimination». The Organometallic HyperTextBook. Interactive Learning Paradigms Inc.
| Control de autoridades |
|
|---|
 Datos: Q2123471
Datos: Q2123471 Multimedia: Oxidative addition / Q2123471
Multimedia: Oxidative addition / Q2123471