Eliminación reductora
La eliminación reductora (también llamada eliminación reductiva) es un paso elemental en la química organometálica en el que el estado de oxidación del centro metálico disminuye, es decir, se reduce, mientras se forma un nuevo enlace covalente entre dos ligandos. Es el reverso microscópico de la adición oxidativa y, a menudo, es una etapa de formación de productos en muchos procesos catalíticos. Dado que la adición oxidativa y la eliminación reductora son reacciones inversas, se aplican los mismos mecanismos para ambos procesos y el equilibrio del producto depende de la termodinámica de ambas direcciones.[1][2]
Información general
La eliminación reductora se ve a menudo en estados de oxidación más altos y puede implicar un cambio de dos electrones en un solo centro metálico (mononuclear) o un cambio de un electrón en cada uno de los dos centros metálicos (binuclear, dinuclear o bimetálico).[1][2]
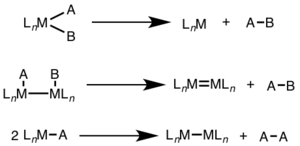
Para la eliminación reductora mononuclear, el estado de oxidación del metal disminuye en dos, mientras que el recuento de electrones d del metal aumenta en dos. Esta vía es común para los metales d8 Ni(II), Pd(II) y Au(III) y los metales d6 Pt(IV), Pd(IV), Ir(III) y Rh(III). Además, la eliminación reductora mononuclear requiere que los grupos que se eliminen estén en cis entre sí en el centro metálico.[3]
Para la eliminación reductora binuclear, el estado de oxidación de cada metal disminuye en uno, mientras que el recuento de electrones d de cada metal aumenta en uno. Este tipo de reactividad se observa generalmente con los metales de primera fila, que prefieren un cambio de una unidad en el estado de oxidación, pero se ha observado tanto en metales de segunda como de tercera fila.[4]
Mecanismos
Al igual que con la adición oxidativa, son posibles varios mecanismos con la eliminación reductora. El mecanismo destacado es una vía concertada, lo que significa que es un estado de transición no polar de tres centros con retención de la estereoquímica. Además, un mecanismo SN2, que procede con la inversión de la estereoquímica, o un mecanismo radical, que procede con la obliteración de la estereoquímica, son otras vías posibles para la eliminación reductiva.[1]
Complejos octaédricos
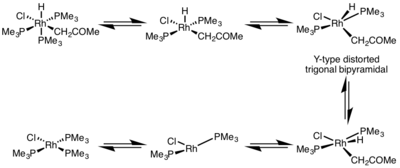
La velocidad de la eliminación reductora está muy influenciada por la geometría del complejo metálico. En los complejos octaédricos, la eliminación reductora puede ser muy lenta desde el centro coordinativamente saturado y, a menudo, la eliminación reductora solo procede a través de un mecanismo disociativo, donde un ligando debe disociarse inicialmente para formar un complejo de cinco coordenadas. Este complejo adopta una estructura bipiramidal trigonal distorsionada de tipo Y donde un ligando dador π está en la posición basal y los dos grupos a eliminar se acercan mucho. Después de la eliminación, se forma un complejo de tres coordenadas en forma de T, que se asociará con un ligando para formar el complejo de cuatro coordenadas plano cuadrado.[5]
Complejos plano-cuadrados
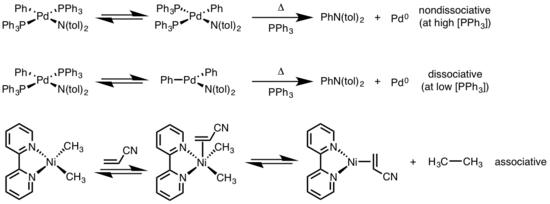
La eliminación reductora de complejos planos cuadrados puede progresar a través de una variedad de mecanismos: disociativo, no disociativo y asociativo. Similar a los complejos octaédricos, un mecanismo disociativo para los complejos planos cuadrados se inicia con la pérdida de un ligando, generando un intermedio de tres coordenadas que se somete a una eliminación reductiva para producir un complejo metálico de una sola coordenada. Para una vía no disociativa, la eliminación reductiva se produce a partir del sistema de cuatro coordenadas para producir un complejo de dos coordenadas. Si los ligandos que se eliminan son trans entre sí, el complejo primero debe someterse a una isomerización de trans a cis antes de eliminarse. En un mecanismo asociativo, un ligando debe asociarse inicialmente con el complejo metálico de cuatro coordenadas para generar un complejo de cinco coordenadas que se somete a una eliminación reductiva análoga al mecanismo de disociación de los complejos octaédricos.[6][7]
Aplicaciones
Cabe decir, para terminar, que esta reacción es muy importante en procesos catalíticos. [8] [9][10]
La eliminación reductora ha encontrado una amplia aplicación en la academia y la industria, siendo las más notables la hidrogenación,[11] el Proceso Monsanto,[12] la hidroformilación,[13] y las reacciones de acoplamiento cruzado.[14] En muchos de estos ciclos catalíticos, la eliminación reductora es el paso de la formación del producto y regeneración del catalizador; sin embargo, en la reacción de Heck[15] y el proceso Wacker,[16] la eliminación reductora está involucrada solo en la regeneración del catalizador, ya que los productos en estas reacciones se forman a través de la beta eliminación.
Referencias
- Crabtree, Robert H. (2014). The Organometallic Chemistry of the Transition Metals (6 edición). Wiley. p. 173. ISBN 978-1-118-13807-6.
- Hartwig, John F. (2010). Organotransition Metal Chemistry, from Bonding to Catalysis. University Science Books. p. 321. ISBN 978-1-891389-53-5.
- Gillie, A.; Stille, J. K. (1980). «Mechanisms of 1,1-Reductive Elimination from Palladium». J. Am. Chem. Soc. 102 (15): 4933-4941. doi:10.1021/ja00535a018.
- Okrasinski, S. J.; Nortom, J. R. (1977). «Mechanism of Reductive Elimination. 2. Control of Dinuclear vs. Mononuclear Elimination of Methane from cis-Hydridomethyltetracarbonylosmium». J. Am. Chem. Soc. 99: 295-297. doi:10.1021/ja00443a076.
- Milstein, D. (1982). «The First Isolated, Stable cis-Hydridoalkylrhodium Complexes and Their reductive Elimination Reaction». J. Am. Chem. Soc. 104 (19): 5227-5228. doi:10.1021/ja00383a039.
- Driver, M. S.; Hartwig, J. F. (1997). «Carbon−Nitrogen-Bond-Forming Reductive Elimination of Arylamines from Palladium(II) Phosphine Complexes». J. Am. Chem. Soc. 119 (35): 8232-8245. doi:10.1021/ja971057x.
- Yamamoto, T.; Yamamoto, A.; Ikeda, S. (1971). «Study of Organo(dipyridyl)nickel Complexes. I. Stability and Activation of the Alkyl-Nickel Bonds of Dialkyl(dipyridyl)nickel by Coordination with Various Substituted Olefins». J. Am. Chem. Soc. 93: 3350. doi:10.1021/ja00743a009.
- Curso organometálicos
- Astruc, Didier (2003). «Principales familias de complejos organometalicos». Química organometálica (escrit). Alvagraf, S.L. 08120 La LLagosta (BARCELONA): Reverté. p. 540. ISBN 84-291-7007-3.
- de Vries, J. G. (2007). The Handbook of Homogeneous Hydrogenation. Wiley. ISBN 978-3-527-31161-3.
- Paulik, F. E.; Roth, J. F. (1968). «Novel Catalysts for the Low-pressure Carbonylation of Methanol to Acetic Acid». Chem. Commun. (24): 1578. doi:10.1039/C1968001578A.
- Ojima, I.; Tsai, C.-H.; Tzamarioudaki, M.; Bonafoux, D. (2004). «The Hydroformylation Reaction». Organic Reactions 56: 1-354. ISBN 0471264180. doi:10.1002/0471264180.or056.01.
- New Trends in Cross-Coupling: Theory and Applications Thomas Colacot (Editor) 2014 ISBN 978-1-84973-896-5
- de Vries, J. G. (2001). «The Heck reaction in the production of fine chemicals». Can. J. Chem. 79 (5–6): 1086-1092. doi:10.1139/v01-033. hdl:11370/31cf3b82-e13a-46e2-a3ba-facb1e2bffbf.
- Dong, J. J.; Browne, W. R.; Feringa, B. L. (2015). «Palladium-Catalyzed anti-Markovnikov Oxidation of Terminal Alkenes». Angew. Chem. Int. Ed. 54 (3): 734-744. PMID 25367376. doi:10.1002/anie.201404856.
| Control de autoridades |
|
|---|
 Datos: Q25325213
Datos: Q25325213