Ciclo de la urea
El ciclo de la urea es el proceso metabólico en el cual se procesan los derivados proteicos y se genera urea como producto final.
Si no se reutilizan para la síntesis de nuevos aminoácidos u otros productos nitrogenados, los grupos amino se canalizan a un único producto final de excreción. La mayoría de especies acuáticas, como por ejemplo los peces óseos, excretan el nitrógeno amínico en forma de amoníaco por lo que se les llama animales amonotélicos; la mayoría de animales terrestres son ureotélicos, excretan el nitrógeno amínico en forma de urea; las aves y también los reptiles son uricotélicos, excretan el nitrógeno amínico en forma de ácido úrico.
En los organismos ureotélicos, el amoníaco depositado en las mitocondrias de los hepatocitos se convierte en urea mediante el ciclo de la urea.
Esta ruta fue descubierta en 1932 por Hans Krebs (quien más tarde también descubriría el ciclo del ácido cítrico) y un estudiante médico asociado, Kurt Henseleit. La producción de urea tiene lugar casi exclusivamente en el hígado y representa el destino de la mayor parte del amoníaco allí canalizado. La urea pasa al torrente sanguíneo y de ahí a los riñones y se excreta en la orina, dando así urea como producto final.
Ciclo de la urea
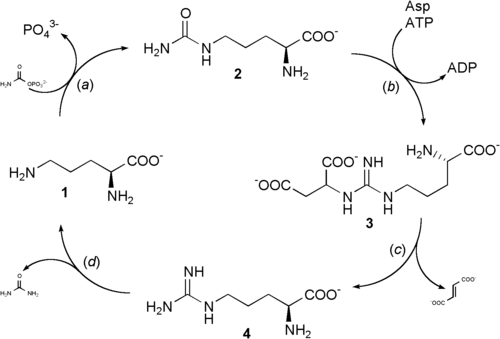
El ciclo de la urea empieza desde el interior de las mitocondrias del hígado, si bien tres de los pasos siguientes tienen lugar en el citosol; por lo tanto, el ciclo abarca dos compartimientos celulares. El ciclo de la urea proviene del amoníaco de la matriz mitocondrial, como resultado de las múltiples rutas descritas. Parte del amoníaco también llega al hígado vía vena porta a partir del intestino en donde se produce por oxidación bacteriana de aminoácidos. Cualquiera sea su origen, el NH4 generado en las mitocondrias hepáticas se utiliza únicamente e inmediatamente junto con el CO2 (en forma de HCO3-) producido por la respiración mitocondrial, generando carbamoil fosfato en la matriz. Esta reacción dependiente de ATP es catalizada por la carbamoil fosfato sintetasa I, la enzima reguladora. La forma mitocondrial de la enzima es distinta de la forma citosólica (II), cumple una función diferente en la síntesis de pirimidinas. El carbamoil fosfato, que puede ser considerado como un donador activado del grupo carbamilo, entra ahora en el ciclo de la urea, que consta de cuatro pasos enzimáticos. En primer lugar, el carbamoil fosfato cede su grupo carbamilo a la ornitina para formar citrulina y libera Pi y tiene lugar a través de un intermedio citrulil-AMP'. La ornitina desempeña pues un papel similar al del oxalacetato en el ciclo del ácido cítrico, aceptando material en cada vuelta del ciclo. La reacción está catalizada por la ornitina transcarbamilasa, y la citrulina formada pasa de la mitocondria al citosol.
El segundo grupo amino, se introduce a partir del aspartato (generado en la mitocondria por transaminación y transportado al citosol) mediante una reacción de condensación entre el grupo amino del aspartato y el grupo ureido (carbonilo) de la citrulina, que forma argininosuccinato. Este tipo de reacción citosólica, catalizada por la argininosuccinato sintetasa, requiere ATP. A continuación, se corta reversiblemente el argininosuccinato por la argininosuccinato liasa, para formar arginina libre y fumarato, que entra en la mitocondria y se une a la reserva de intermedios del ciclo del ácido cítrico. En la última reacción del ciclo de la urea, la enzima citosólica arginasa corta la arginina dando urea y ornitina. La ornitina es transportada a la mitocondria para iniciar otra vuelta del ciclo de la urea.
Las enzimas de muchas rutas metabólicas están agrupados. El producto de una enzima se canaliza directamente al siguiente enzima de la vía. En el ciclo de la urea, los enzimas mitocondriales y citosólicos parecen estar agrupados de esta forma. La citrulina transportada al exterior de la mitocondria no se diluye en la reserva general de metabolitos del citosol sino que pasa directamente al centro activo de la argininosuccinato sintetasa. Esta canalización entre enzimas continúa para el argininosuccinato, arginina y ornitina. Sólo se libera la urea a la reserva general de metabolitos del citosol.
| Pasos | Reactantes | Productos | Catalizado por | Localización |
|---|---|---|---|---|
| 1 | 2ATP + HCO3− + NH3+ | carbamoil fosfato + 2ADP + Pi | CPS1 | mitocondria |
| 2 | carbamoil fosfato + ornitina | citrulina + Pi | OTC | mitocondria |
| 3 | citrulina + aspartato + ATP | argininosuccinato + AMP + PPi | ASS | citosol |
| 4 | argininosuccinato | Arg + fumarato | ASL | citosol |
| 5 | Arg + H2O | ornitina + urea | ARG1 | citosol |
- Las reacciones del ciclo de la urea
1 L-ornitina
2 carbamil fosfato
3 L-citrulina
4 argininosuccinato
5 fumarato
6 L-arginina
7 urea
L-Asp L-aspartato
CPS-1 carbamil fosfato sintetasa I
OTC Ornitina transcarbamilasa
ASS argininosuccinato sintetasa
ASL argininosuccinato liasa
ARG1|Arginasa1

Relación con el ciclo de Krebs
Dado que el fumarato producido en la reacción de la argininosuccinato liasa es también un intermediario del ciclo del ácido cítrico, los ciclos están, en principio, interconectados –en un proceso conocido como el “doble ciclo de Krebs”-. Sin embargo, cada ciclo puede funcionar de manera independiente y la comunicación entre ellos depende del transporte de intermedios clave entre la mitocondria y el citosol. Varias enzimas del ciclo del ácido cítrico, incluyendo la fumarasa (fumarato hidratasa y la malato deshidrogenasa) también están presentes como isozimas en el citosol. El fumarato generado en la síntesis citosólica de arginina puede, por tanto, convertirse en malato y a continuación en oxalacetato en el citosol, y estos intermedios pueden seguir siendo metabolizados en el citosol o ser transportados a las mitocondrias para su utilización en el ciclo del ácido cítrico.
El aspartato formado en las mitocondrias por transaminación entre oxalacetato y glutamato puede ser transportado al citosol, en donde actúa como donador de nitrógeno en la reacción del ciclo de la urea catalizada por la argininosuccinato sintetasa. Estas reacciones, que constituyen la desviación del aspartato-argininosuccinato, proporcionan vínculos metabólicos entre las rutas separadas por las que se procesan los grupos amino y los esqueletos carbonados de los aminoácidos.
Regulación del ciclo de la urea
El flujo de nitrógeno a través del ciclo de la urea varía con la dieta de un organismo. Cuando la dieta es mayoritariamente proteína, la utilización de los esqueletos carbonados de los aminoácidos como combustible da lugar a la producción de mucha urea a partir del exceso de grupos amino. Durante la inanición prolongada, en la que la degradación de proteína muscular empieza a suministrar gran parte de la energía metabólica del organismo, también aumenta sustancialmente la producción de urea.
Estos cambios en la demanda de actividad del ciclo de la urea se consiguen a largo plazo mediante la regulación de las velocidades de síntesis de los cuatro enzimas del ciclo de la urea y de la carbamil fosfato sintetizada en el hígado. Las cinco enzimas se sintetizan a velocidades más elevadas durante la inanición o en los animales con dietas muy ricas en proteínas que en animales bien alimentados con dietas que contienen principalmente glúcidos y grasas. Los animales con dietas carentes de proteínas producen niveles más bajos de los enzimas del ciclo de la urea.
En una escala de tiempo más corta, la regulación alostérica de al menos una enzima clave ajusta el flujo a través del ciclo de la urea. La primera enzima de la ruta, la carbamil fosfato sintetasa I, está activada alostéricamente por el N-acetilglutamato, que es sintetizado a partir del acetil-CoA y glutamato por la N-acetilglutamato sintasa. Este enzima cataliza el primer paso de la síntesis de novo de la arginina a partir de glutamato en plantas y microorganismos. Los mamíferos, sin embargo, tienen actividad N-acetilglutamato sintasa en el hígado, pero carecen del resto de enzimas necesarias para convertir glutamato en arginina. Por tanto, el uso de N-acetilglutamato para activar un paso en el ciclo de la urea resulta enigmático.
Enfermedades
Las personas con defectos genéticos en cualquiera de los enzimas que intervienen en la formación de urea no pueden tolerar una dieta rica en proteínas. Los aminoácidos ingeridos en exceso con respecto a los requerimientos diarios mínimos para la síntesis de proteínas se desaminan en el hígado, produciendo amoníaco libre que no puede ser convertido en urea y exportado a la sangre, y el amoníaco es muy tóxico. Los humanos, sin embargo, no podemos vivir de una dieta carente de proteína. Somos incapaces de sintetizar la mitad de los 20 aminoácidos estándar, y estos aminoácidos esenciales deben ser suministrados en la dieta.
A los pacientes con defectos en el ciclo de la urea se les aplican diversos tratamientos. La administración cuidadosa de los ácidos aromáticos benzenoato o fenilacetato en la dieta puede ayudar a reducir los niveles de amoníaco en la sangre. El benzenoato se convierte en benzenoil-CoA, que se combina con la glicina formando hipurato. La glicina utilizada en esta reacción debe ser regenerada, y así el amoníaco se utiliza en la reacción de la glicina sintasa. El fenilacetato se combina con glutamina para formar fenilacetilglutamina, y la subsiguiente síntesis de glutamina sintasa ayuda a eliminar amoníaco. Tanto el hipurato como la fenilacetilglutamina son compuestos no tóxicos que se excretan en la orina.
Otras terapias son más específicas de una determinada deficiencia enzimática. La deficiencia de N-acetilglutamato sintetasa tiene como resultado la ausencia del activador normal de la carbamil fosfato sintetasa I. Este estado puede tratarse administrando carbamil glutamato, un análogo del N-acetilglutamato que es efectivo como activador de la carbamil fosfato sintetasa I. Para las deficiencias en ornitina transcarbamilasa, argininosuccinato sintetasa y argininosuccinato liasa, es útil sumplementar la dieta con arginina. Muchos de estos tratamientos deben ir acompañados de un control estricto de la dieta y de suplementos de los aminoácidos esenciales. En los pocos casos de deficiencia de arginasa, la arginina, el sustrato del enzima defectivo, debe excluirse en la dieta.
Ecología
La síntesis de urea no es la única ruta, ni siquiera la más común, para excretar el amoníaco. La base para las diferentes formas moleculares en que se excretan los grupos amino se encuentra en la anatomía y en la fisiología de los organismos en relación con su hábitat natural. Las bacterias y los protozoos simplemente liberan el amoníaco en su entorno acuoso, en el que se diluye, con lo que se convierte en una forma inocua. En los peces óseos (animales amonotélicos), el hígado es el lugar principal del catabolismo de los aminoácidos. El amonio producido por transdesaminación se libera simplemente del hígado a la sangre para su transporte a las branquias, y es rápidamente eliminado de la sangre a medida que el agua pasa a través de las branquias. Así pues, los peces óseos no requieren un complejo sistema urinario.
En las aves y reptiles, la disponibilidad de agua para el proceso excretor constituye una consideración especialmente importante. La excreción de urea en la orina requiere la excreción simultánea de una cantidad relativamente grande de agua; el peso constituiría un impedimento para las aves en vuelo, mientras que los reptiles que viven en ambientes áridos no pueden tener agua sobrante. Estos animales convierten el nitrógeno amínico en purinas, las cuales se catabolizan a ácido úrico, un compuesto bastante insoluble que se excreta en las heces en forma de masa semisólida de cristales de ácido úrico. Para tener la ventaja de excretar el nitrógeno amínico en esta forma, las aves y reptiles llevan a cabo un trabajo metabólico considerable; la ruta desde los grupos amino de los aminoácidos a las purinas y al ácido úrico es un proceso complejo y que requiere energía.
Propiedades de la urea
•Molécula pequeña, sin carga.
•Difusible
•Soluble
•Atóxica
•El 50% de su peso es nitrógeno.
•Permite la eliminación de 2 productos de desecho: CO2 y NH3
•Valores Normales: 25-30 gr de urea diarios
Véase también
Galería
 Ciclo de la urea
Ciclo de la urea
Enlaces externos
- http://homepage.ufp.pt/pedros/bq/urea.htm (The chemical logic behind the urea cycle)
- http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=bnchm.figgrp.3102 (Basic Neurochemistry)
| Control de autoridades |
|
|---|
 Datos: Q595994
Datos: Q595994 Multimedia: Urea cycle / Q595994
Multimedia: Urea cycle / Q595994