Glomerulonefritis rápidamente progresiva
La glomerulonefritis rápidamente progresiva o extracapilar es un síndrome renal que si no se trata progresa con rapidez a una insuficiencia renal aguda y a la muerte del paciente en cuestión de meses. En el cincuenta por ciento de los casos esta glomerulonefritis se asocia con otra enfermedad de base, como por ejemplo el síndrome de Goodpasture, el lupus eritematoso sistémico o la granulomatosis de Wegener. Los casos restantes son idiopáticos, es decir, se ignora la causa de la aparición de la enfermedad. De todos modos, independientemente de la etiología principal, la glomerulonefritis rápidamente progresiva implica una serie de lesiones del glomérulo del riñón y la presencia de las típicas cicatrices con forma de semiluna en más del cincuenta por ciento de las unidades glomerulares.[1] En ocasiones, debido a esta característica histológica, la enfermedad recibe el nombre de glomerulonefritis semilunar.[1]
| Glomerulonefritis rápidamente progresiva | ||
|---|---|---|
.jpg.webp) Imagen histopatológica de una muestra de glomerulonefritis con semilunas obtenida de un paciente con glomerulonefritis rápidamente progresiva positiva para anticuerpos anti-MPO-ANCA. Tinción con hematoxilina y eosina. | ||
| Especialidad | nefrología | |
Clasificación
La glomerulonefritis rápidamente progresiva se clasifica en tres tipos, todos asociados con daño glomerular mediado por el sistema inmunitario. En el tipo I los anticuerpos se dirigen directamente contra la membrana basal glomerular, el tipo II se caracteriza por el depósito de complejos inmunes sobre el glomérulo y en el tipo III hay anticuerpos contra el citoplasma de los neutrófilos o ANCA (del inglés antineutrophil cytoplasm antibodies).
Tipo I
Este tipo, que explica alrededor del treinta por ciento de los casos de glomerulonefritis rápidamente progresiva, se caracteriza por la presencia de anticuerpos dirigidos contra la membrana basal del glomérulo, por lo que también se llama glomerulonefritis antimembrana basal glomerular. Los anticuerpos mencionados tienen especificidad por determinadas proteínas de la membrana basal, a saber, el colágeno de tipo IV, específicamente la región no colagenosa de su cadena α3.[2]
Además de los anticuerpos antimembrana basal glomerular, en algunos casos de glomerulonefritis rápidamente progresiva o extracapilar de tipo I se detectan anticuerpos dirigidos contra la membrana basal de los alvéolos del pulmón, lo que produce el síndrome de Goodpasture. Sin embargo, en la mayoría de los pacientes con enfermedad de tipo I solo se detectan anticuerpos antimembrana basal glomerular, los que se consideran idiopáticos.[2]
Tipo II
La glomerulonefritis rápidamente progresiva causada por el depósito de complejos inmunes representa el veinticinco por ciento de los casos y se clasifica como de tipo II. Cualquier enfermedad por complejos inmunes que afecte el glomérulo de manera importante tendrá el potencial de convertirse en una glomerulonefritis rápidamente progresiva. Las enfermedades con ese potencial incluyen lupus eritematoso, glomerulonefritis posinfecciosa, púrpura de Schönlein-Henoch y nefropatía por IgA.[2]
Tipo III
La glomerulonefritis rápidamente progresiva cuasiinmune, o de tipo III, explica el cincuenta y cinco por ciento de los casos y se caracteriza por la ausencia de depósitos de complejos inmunes y de anticuerpos antimembrana basal glomerular.[3] En este caso el glomérulo se daña de manera aún no identificada, probablemente por la activación de neutrófilos en respuesta a la presencia de ANCA. La glomerulonefritis rápidamente progresiva de tipo III puede ser de origen glomerular (primaria o idiopática) o estar asociada con una enfermedad sistémica (secundaria). La mayor parte de los casos son secundarios y la enfermedad sistémica es una vasculitis asociada con ANCA, como la granulomatosis de Wegener, la poliangitis microscópica o el síndrome de Churg-Strauss.[2][nota 1]
Historia
En 1914 Volhard y Fahr reconocieron por primera vez la asociación entre la presencia de las semilunas y el curso clínico desfavorable de estas glomerulopatías y las denominaron glomerulonefritis extracapilares; más tarde, en 1942, Ellis les dio el nombre de glomerulonefritis rápidamente progresivas. Otras denominaciones propuestas han sido glomerulonefritis maligna, glomerulonefritis aguda necrosante y glomerulonefritis semilunar.[4] En 1933 Masugi, un anatomopatólogo japonés, describió la enfermedad antimembrana basal glomerular experimental como una nefritis nefrotóxica.[5]
Epidemiología
La glomerulonefritis rápidamente progresiva puede afectar a personas de cualquier edad pero la de tipo I es más frecuente en pacientes jóvenes y la de tipo III predomina en ancianos. Ambos sexos son afectados por igual. En el setenta por ciento de los pacientes la glomerulonefritis extracapilar se presenta como una afección renal primaria o idiopática mientras que en el treinta por ciento restante es secundaria a un gran número de enfermedades sistémicas, infecciosas, autoinmunitarias o neoplásicas.[4] En España esta enfermedad explica algo más del cinco por ciento de las biopsias renales realizadas en adultos y el nueve por ciento de todas las glomerulonefritis primarias detectadas por biopsia.[6][7]
La enfermedad antimembrana basal glomerular, una glomerulonefritis rara pero bien caracterizada[8] y una causa infrecuente de insuficiencia renal aguda, con una incidencia de solo 0,5-1 caso/millón/año,[9] representa del uno al cinco por ciento de todos los tipos de glomerulonefritis y es la causa del diez al veinte por ciento de los casos de glomerulonefritis semilunar.[10]
.jpg.webp)
Según otro autor,[11] la glomerulonefritis rápidamente progresiva o semilunar puede presentarse a cualquier edad pero es infrecuente en los niños, predomina en los hombres (con una relación de 2:1) y su frecuencia es menor en la raza negra. Además, según publicaciones de distintos países (Francia, Estados Unidos, China, la India y África), explica del dos al cinco por ciento de los hallazgos de biopsia. Por último, se presenta como insuficiencia renal aguda en el cinco al seis por ciento de los adultos, en el doce al quince por ciento de los niños y en el veinte al treinta y cuatro por ciento de los casos atípicos.
Etiología
En cerca del cuarenta por ciento de los casos la glomerulonefritis rápidamente progresiva (síndrome nefrítico rápidamente progresivo) forma parte de un trastorno que afecta a otros órganos, además de los riñones. En el sesenta por ciento de los casos con compromiso de los riñones alrededor de un tercio parece ser causado por anticuerpos que atacan a los glomérulos; de ese tercio, cerca de la mitad se debe a causas desconocidas y el resto es provocado por el depósito renal de anticuerpos y antígenos que se han formado en otra parte del cuerpo (enfermedad por complejos inmunes).[12] Se ignora la causa por la que el organismo produce anticuerpos contra sus propios glomérulos. La producción de estos anticuerpos perjudiciales puede estar relacionada con infecciones virales o con trastornos autoinmunitarios como el lupus eritematoso sistémico. En algunas personas que desarrollan anticuerpos contra sus glomérulos los anticuerpos también reaccionan contra los alvéolos pulmonares y producen el síndrome de Goodpasture, un proceso que compromete los pulmones y los riñones. Los hidrocarburos como el etilenglicol, el tetracloruro de carbono, el cloroformo y el tolueno pueden lesionar los glomérulos pero no provocan una reacción inmunitaria ni la generación de anticuerpos.[12]
Patogenia
La producción de interleucina I, la interacción de las moléculas de adhesión y la proliferación local de los macrófagos son factores que intervienen en la génesis de las lesiones.[4]
En la enfermedad mediada por anticuerpos dirigidos de forma específica contra antígenos de la membrana basal glomerular o de tipo I los estudios con inmunofluorescencia revelan la presencia de depósitos lineales y continuos de inmunoglobulinas, principalmente IgG, a todo lo largo de la membrana.[4]
Cuando esta glomerulonefritis se asocia con hemorragia pulmonar constituye el síndrome de Goodpasture, dado que se ha demostrado una reacción cruzada de los anticuerpos antimembrana basal glomerular con las membranas basales de los alvéolos pulmonares y que ambas membranas basales comparten estructuras antigénicas similares. Alrededor del veinte por ciento de las glomerulonefritis rápidamente progresivas pertenecen a este grupo.[4]
En la enfermedad de tipo II, es decir en la mediada por inmunocomplejos circulantes o formados in situ a nivel glomerular, los estudios con inmunofluorescencia demuestran la presencia de depósitos granulares y discontinuos de inmunoglobulinas acompañados habitualmente por complemento. Dentro de este grupo se encuentra del treinta al cuarenta por ciento de las glomerulonefritis rápidamente progresivas, que en la mayor parte de los casos son secundarias a una enfermedad sistémica como lupus eritematoso diseminado, crioglobulinemia, púrpura de Schönlein-Henoch y las asociadas con algunas infecciones.[4]
Por último, el cuarenta por ciento de las glomerulonefritis rápidamente progresivas corresponden a la categoría denominada glomerulonefritis cuasiinmune o sin depósitos inmunes o de tipo III. En un gran número de pacientes los estudios serológicos detectan la presencia de ANCA.
El primer paso es el ataque inmunitario de la membrana basal glomerular con aparición de auténticos "orificios" a través de los cuales van a pasar distintos componentes de la sangre que luego ingresarán al espacio de Bowman. Diversos factores plasmáticos, entre los que se destaca la fibrina, actuarán como el estímulo quimiotáctico y proliferativo que dará lugar a la formación de las semilunas. Esta evolución tan característica determina las diferentes morfologías de las semilunas, que según los estudios con microscopia óptica son celulares, fibrocelulares o fibrosas. El resultado final es la evolución a un cuadro de fibrosis progresiva y esclerosis glomerular.[13] De acuerdo con lo informado por los autores de varios trabajos en este proceso intervienen moléculas de adhesión celular, interleucinas, integrinas y factores de crecimiento.[14]

En el riñón una lámina basal doble, la membrana basal glomerular, separa el epitelio que reviste el espacio urinario del endotelio que tapiza los capilares circundantes llenos de sangre. Los defectos de esa estructura, que está a cargo de la ultrafiltración de la sangre y de la formación inicial de la orina, pueden causar insuficiencia renal. Por ejemplo, las mutaciones que alteran el dominio globular C-terminal de las cadenas α3 del colágeno de tipo IV se asocian con insuficiencia renal progresiva. En el síndrome de Goodpasture, una enfermedad autoinmunitaria bastante rara en la que el organismo se “autoataca”, los anticuerpos se unen a las cadenas α3 del colágeno de tipo IV situadas en la membrana basal glomerular y en los pulmones y esa unión desencadena una respuesta inmunitaria que provoca daño celular, el que se traduce en insuficiencia renal progresiva y hemorragia pulmonar.[7] La glomerulonefritis rápidamente progresiva constituye la forma más agresiva de glomerulonefritis, con la frecuencia más alta de insuficiencia renal y formación de semilunas en el momento del diagnóstico.[15]
Formación de las semilunas
Pese a la gran variedad de enfermedades que causan glomerulonefritis rápidamente progresiva, todos los tipos se caracterizan por daño glomerular y formación de semilunas. El daño más grave y la ruptura de la membrana basal glomerular conducen a la pérdida de proteínas plasmáticas. De estas proteínas, se piensa que la fibrina es la que más contribuye a la formación de semilunas. Las células epiteliales que delimitan la cápsula de Bowman responden al estímulo proliferante de la fibrina. También puede haber infiltración de leucocitos como los monocitos y los macrófagos, los que también pueden proliferar bajo la influencia de la fibrina. Estas células proliferantes rodean al glomérulo y lo comprimen por lo que aparece la cicatriz con forma de semiluna que es visible con el microscopio en el material obtenido de una biopsia renal.[2]

Aunque algo arbitrario, actualmente se considera que debe de haber al menos cincuenta por ciento de glomérulos con semilunas para llamar a esta enfermedad glomerulonefritis extracapilar (crescentic).[15]
Anatomía patológica
En la biopsia de estos pacientes se halla una importante proliferación de las células epiteliales de la cápsula de Bowman, con formación de semilunas en los glomérulos.[4] La biopsia renal confirma la presencia de una glomerulonefritis con semilunas epiteliales. Entre las características histopatológicas figura la infiltración del espacio urinario por células mononucleares asociada con la proliferación del epitelio parietal de la cápsula de Bowman o la proliferación extracapilar con formación de semilunas que pueden afectar del treinta al cien por ciento de los glomérulos. En estos se ve un engrosamiento de la cápsula de Bowman con formación de semilunas. También puede haber fibrosis parcial y sinequia del penacho glomerular a la cápsula.[4] Las semilunas, que pueden ser segmentarias o circunferenciales, ocupan todo el espacio de Bowman y ahogan el ovillo capilar, según el estado evolutivo pueden clasificarse en celulares, fibrocelulares o fibrosas y representan fases progresivas del mismo proceso.[4] En la glomerulonefritis rápidamente progresiva de tipo I hay positividad lineal sobre todo para IgG pero después también para C3. Dos tercios de las glomerulonefritis semilunares de este tipo corresponden al síndrome de Goodpasture y el otro tercio carece de manifestaciones pulmonares (síndrome de Goodpasture sin compromiso pulmonar). La lesión glomerular es similar en ambos grupos y se produce por un mecanismo inmunitario antimembrana basal glomerular. Como ya se dijo, el factor desencadenante es un defecto de un componente proteico de la cadena α3 del colágeno de tipo IV.[5] En la enfermedad de tipo II hay positividad granular o nodular o de ambas formas, difusa y global, debido a depósitos de complejos inmunes que con el microscopio electrónico se ven como depósitos densos. Este tipo por lo general corresponde a variedades de la glomerulonefritis aguda difusa, de la glomerulonefritis mesangiocapilar y, menos a menudo, de la enfermedad de Berger.[5]
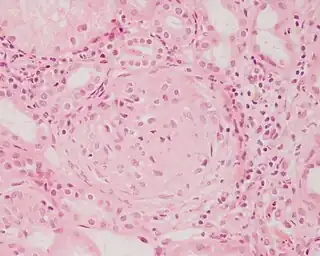
Por último, en la glomerulonefritis semilunar de tipo III el examen con inmunofluorescencia es negativo y los hallazgos detectados con el microscopio electrónico son similares a los de la enfermedad de tipo I (sin depósitos densos). Si bien se ignora la patogenia de esta lesión glomerular, se piensa que en una buena cantidad de casos la causa de la ruptura de capilares glomerulares y de la proliferación celular glomérulo-capsular sería una vasculitis o una angiopatía. Este cuadro se observa con mayor frecuencia en adultos mayores.[5]

Cuadro clínico

En casi todos los pacientes con glomerulonefritis rápidamente progresiva la enfermedad se inicia con hematuria, proteinuria y, ocasionalmente, hipertensión arterial y edema. El cuadro clínico es compatible con un síndrome nefrítico, aunque el grado de proteinuria a veces puede ser mayor de 3 g/24 h, un intervalo generalmente asociado con el síndrome nefrótico. En la enfermedad que evoluciona sin tratamiento se produce una disminución del volumen de orina (oliguria), fenómeno que se vincula con deterioro de la función renal e implica un pronóstico desfavorable.[2]
En el cincuenta por ciento de los casos se observan síntomas prodrómicos que semejan una infección viral con fiebre, artralgias, mialgias, dolor abdominal y lumbalgia, aunque no se ha demostrado su asociación con ninguna virosis específica.[4] El comienzo, insidioso o agudo, se caracteriza por la instauración de una insuficiencia renal rápidamente progresiva que evoluciona en semanas o pocos meses hacia una insuficiencia renal grave o terminal. El deterioro de la función renal se considera rápidamente progresivo si los niveles séricos de creatinina se duplican en un lapso menor de dos meses.[4]
Cuando la enfermedad de base es el síndrome de Goodpasture o una vasculitis que compromete el pulmón, como la granulomatosis de Wegener, los pulmones y las vías respiratorias altas pueden estar dañados y la glomerulonefritis puede acompañarse de tos, dificultad respiratoria y pérdida del apetito.
El examen de orina revela la presencia de eritrocitos dismórficos, cilindros hemáticos y un grado variable de proteinuria.
Diagnóstico
El análisis serológico puede ayudar a establecer el diagnóstico correcto de la causa primaria. La presencia de anticuerpos antimembrana basal glomerular sugiere glomerulonefritis rápidamente progresiva de tipo I, los anticuerpos antinucleares o ANA apoyan el diagnóstico de lupus y la glomerulonefritis rápidamente progresiva de tipos II y III así como la idiopática pueden verse en asociación con una prueba positiva para ANCA.
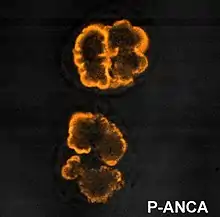
El diagnóstico se establece sobre la base de la sospecha clínica en un paciente con disminución rápida y progresiva del índice de filtración glomerular (IFG) acompañada de proteinuria, por lo general no nefrótica, y hematuria casi siempre microscópica con eritrocitos dismórficos y cilindros granulosos-hemáticos en el sedimento. Este cuadro suele ser precedido por síntomas inespecíficos, un cuadro seudogripal o un síndrome constitucional (cansancio, inapetencia y pérdida de peso). Con frecuencia existe oliguria. El hallazgo de anticuerpos antimembrana basal glomerular, complejos inmunes circulantes o ANCA apoya el diagnóstico, que solo se podrá establecer de forma definitiva mediante el estudio inmunohistológico. Los marcadores inmunológicos orientan el diagnóstico de los distintos tipos de glomerulonefritis rápidamente progresiva pero no hay que olvidar que su sensibilidad y su especificidad no son absolutas y que pueden coexistir en un mismo paciente. En el treinta por ciento de los casos de enfermedad por anticuerpos antimembrana basal glomerular se detecta una positividad simultánea para ANCA que parece contribuir a la lesión de la membrana. Se han descrito solapamientos entre distintas formas de glomerulonefritis rápidamente progresiva.[6]

En alrededor del trece por ciento de los pacientes con enfermedad antimembrana basal glomerular los anticuerpos no son detectables con las técnicas serológicas convencionales[16] por lo que algunos autores recomiendan que el diagnóstico de esta entidad sea considerado dentro del contexto clínico.[17] Algunos de estos pacientes podrían tener anticuerpos dirigidos contra otros antígenos en la membrana basal glomerular, como la entactina.[15][17]
Diagnóstico diferencial
De los depósitos inmunes granulares
Si en la biopsia o en la inmunofluorescencia de las muestras obtenidas de un paciente con glomerulonefritis semilunar se detectan depósitos inmunes granulares se debe pensar en la posibilidad de una infección. De acuerdo con el autor de un trabajo en Maracaibo[11] (Venezuela) la primera opción es la glomerulonefritis posestreptocócica pero deben tenerse presentes otras infecciones, como la endocarditis bacteriana y los abscesos viscerales.
Además hay que recordar que las enfermedades sistémicas causadas por complejos inmunes también producen depósitos granulares. Las más importantes de esas enfermedades son el lupus eritematoso sistémico, la púrpura de Schönlein-Henoch y, si bien menos frecuentes, las gammapatías monoclonales.[11]
Algunas glomerulonefritis crónicas de etiología no bien definida se asocian con depósitos granulares, como la glomerulonefritis membranoproliferativa, la glomerulonefritis membranosa con evolución a semilunar y la nefropatía por IgA, entre otras etiologías.
Las glomerulonefritis que en la biopsia muestran ausencia de depósitos inmunes, o la presencia de muy pocos, son positivas para ANCA en el ochenta por ciento de los casos y ante esa situación se debe plantear la posibilidad de vasculitis idiopática, poliarteritis microscópica, granulomatosis de Wegener y otras entidades menos frecuentes.[11]
Si se encuentra una inmunofluorescencia lineal lo primero que se debe considerar es la posibilidad de síndrome de Goodpasture o enfermedad antimembrana basal glomerular, aunque esos resultados también se pueden deber a fármacos como la penicilamina, la hidralazina y el enalapril.[11]
Hay glomerulonefritis con semilunas en enfermedades malignas como el linfoma y el carcinoma y en enfermedades debidas a silicosis y solventes orgánicos. También, en algunos casos, se ha sugerido la asociación con mordedura de serpiente o deficiencia de alfa 1-antitripsina.[11]
Número de semilunas y patología de base
Las investigaciones realizadas sobre la glomerulonefritis con semilunas han demostrado que existe una relación entre la presencia de las semilunas y algunas enfermedades. Por ejemplo, en la biopsia renal del noventa y cinco por ciento de los pacientes con enfermedad antimembrana basal glomerular o síndrome de Goodpasture con expresión pulmonar se observa al menos una semiluna; en cambio, solo el cincuenta por ciento de los pacientes con lupus y el veinticinco por ciento de los pacientes con nefropatía por IgA, nefritis posestreptocócica o glomerulonefritis membranoproliferativa presentan glomerulonefritis semilunar de tipo II o causada por complejos inmunes.[11]
Las semilunas están presentes en el noventa y cinco por ciento de los casos de glomerulonefritis asociadas con vasculitis, en el cien por ciento de los casos de glomerulonefritis cuasiinmune y en el noventa por ciento de los casos de glomerulonefritis microscópica con poliarteritis. Si la biopsia renal no demuestra la presencia de semilunas es raro que se trate de glomerulonefritis asociada con ANCA, con el síndrome de Goodpasture o con enfermedad antimembrana basal glomerular.[11]
Cuando se analiza el caso de un paciente con diagnóstico de glomerulonefritis semilunar rápidamente progresiva ciertos hallazgos serológicos pueden servir de ayuda. Se puede detectar disminución del complemento sérico en pacientes con lupus (que además son anti-ADN positivos), en pacientes con glomerulonefritis posestreptocócica (que tienen un nivel alto de anticuerpos antiestreptocócicos circulantes), en pacientes con crioglobulinemia (que también tienen crioglobulinas) y en pacientes con glomerulonefritis membranoproliferativa de tipos I y II (que además presentan el factor nefrítico depresor del complemento C3).[11]
Si el paciente tiene un complemento sérico normal se puede tratar de una enfermedad antimembrana basal glomerular, en la que es posible encontrar anticuerpos antimembrana basal circulantes, o de una vasculitis, que se asocia con anticuerpos ANCA circulantes.[11]
Enfermedad antimembrana basal
Si la nefritis semilunar se asocia con depósitos lineales se trata de una enfermedad antimembrana basal.[11] La consideración clínica más importante en el momento de formular el diagnóstico de una glomerulonefritis antimembrana basal glomerular con semilunas es la creatinina sérica. Algunos estudios demuestran que si la concentración de creatinina es menor de 7 mg/dl se puede intentar un tratamiento con ciclofosfamida, prednisona y plasmaféresis, pero si es mayor de ese nivel es mejor optar por un tratamiento conservador y diálisis, dado que con otros tratamientos la mortalidad es más elevada. Si el paciente presenta depósitos granulares en la inmunofluorescencia la consideración diagnóstica más importante es si existe o no sintomatología sistémica asociada.[11]
Si no hay síntomas sistémicos la primera posibilidad diagnóstica es la glomerulonefritis membranoproliferativa, en cuyo caso no existe un tratamiento específico de modo que se debe esperar la evolución e implementar las medidas tradicionales de sostén. En segundo lugar puede tratarse de una glomerulonefritis posestreptocócica sin síntomas sistémicos.[11]
Si hay síntomas sistémicos se debe pensar en las glomerulonefritis asociadas con infecciones, en las que el tratamiento antibiótico es una opción terapéutica importante, o en el lupus eritematoso sistémico, que exige tratamiento con esteroides o ciclofosfamida.[11]
Con ausencia de depósitos inmunes
Si el paciente no presenta depósitos inmunes también habrá que preguntarse si hay o no síntomas sistémicos porque en ausencia de síntomas el diagnóstico más probable es el de una glomerulonefritis rápidamente progresiva idiopática, que muchas veces mejora con la administración de pulsos de esteroides.[11]
Si existen síntomas sistémicos es casi seguro que se trata de una vasculitis, pero una vasculitis sin síntomas sistémicos es rara. En este caso el tratamiento específico consiste en ciclofosfamida, prednisona o plasmaféresis.[11]
Tratamiento
Esta forma de glomerulonefritis, que por fortuna es poco frecuente, se caracteriza por su comienzo insidioso o abrupto y su evolución rápida –en semanas o meses– a la insuficiencia renal terminal si su curso no se modifica mediante la utilización de un tratamiento enérgico (disponible en la actualidad).[4]
Ese tratamiento, que depende de la enfermedad de base (por ejemplo, la plasmaféresis, los corticosteroides y los medicamentos citotóxicos pueden aliviar el síndrome de Goodpasture, una de las causas de la glomerulonefritis rápidamente progresiva de tipo I), deberá ser siempre individualizado y sin olvidar la posibilidad de recuperación en función de los criterios pronósticos. A pesar de un tratamiento temprano, muchos de los pacientes con esta enfermedad requerirán hemodiálisis y posiblemente un trasplante de riñón.
En las formas asociadas con anticuerpos antimembrana basal glomerular o con hemorragia pulmonar se administran en forma conjunta esteroides y ciclofosfamida (o azatioprina como segunda opción), con plasmaféresis. En el resto de los casos el uso de la plasmaféresis resulta más controvertido.[18]
Si tras dos semanas de tratamiento intensivo no se observa recuperación de la función renal habrá que replantear la situación, evaluar la supresión del tratamiento e incluir al paciente en un programa de diálisis. En los casos en los que la enfermedad aparece como complicación de otras glomerulonefritis primarias el pronóstico es malo y con escasa posibilidad de recuperación, fundamentalmente en ancianos, por lo que se deberá plantear un tratamiento conservador, sin la inmunosupresión. Con frecuencia hay otro tipo de patología asociada, como por ejemplo una neoplasia. En las formas secundarias el tratamiento será siempre el de la enfermedad de base.[6]
El empleo de ANCA y anticuerpos antimembrana basal glomerular como marcadores de actividad inmunitaria es muy útil en el seguimiento evolutivo y para fijar las pautas de tratamiento de estas enfermedades.[6]
La posibilidad de recidiva postrasplante es real en los tres tipos de glomerulonefritis extracapilar pero fundamentalmente en la de tipo I, de modo que hay que esperar un mínimo de seis meses desde el episodio inicial antes de incluir al paciente en lista y asegurarse de la negatividad del suero para los anticuerpos y los inmunocomplejos.[6]
Ante el riesgo de infección por Pneumocystis se debe asociar profilaxis con trimetoprima-sulfametoxazol.[19]
Se están ensayando otras formas de tratamiento, como por ejemplo el uso de globulinas antilinfocíticas, anticuerpos monoclonales, técnicas de inmunoadsorción y aféresis de linfocitos,[20] pero la experiencia clínica todavía es escasa.
Otros regímenes terapéuticos que se están probando consisten en la asociación de prednisona oral con ciclofosfamida y azatioprina y la utilización de desoxipergualina,[4][21] un agente inmunosupresor que disminuye la infiltración de los macrófagos y la producción del factor de necrosis tumoral.[4]
Pronóstico
La glomerulonefritis rápidamente progresiva constituye una auténtica urgencia médica, con un pronóstico pésimo y evolución a insuficiencia renal terminal en cuestión de días o a lo sumo de algunas semanas (en general en un plazo inferior a tres meses) si no media un tratamiento adecuado.[6] La evolución a insuficiencia renal terminal es más rápida en la forma de tipo I, en la de tipo II depende sobre todo de la enfermedad de base y puede resolverse espontáneamente y en la de tipo III el pronóstico dependerá de la rapidez con la que se establezca el tratamiento. En términos globales, alrededor del ochenta por ciento de los casos de glomerulonefritis rápidamente progresiva requieren diálisis.
Los factores de mal pronóstico incluyen insuficiencia renal avanzada en el momento del diagnóstico (creatinina > de 6 mg/dl), oligoanuria, riñones disminuidos de tamaño en la ecografía, semilunas de tipo fibroso, esclerosis glomerular en más del veinticinco por ciento de los glomérulos, atrofia tubular extensa y marcada fibrosis intersticial.[6] El valor de la extensión de las semilunas es más controvertido. El porcentaje de glomérulos con semilunas tiene una importancia pronóstica más limitada. La disrupción de la cápsula de Bowman parece desempeñar un papel en la transformación fibrosa de las semilunas y por consiguiente en el pronóstico. También pueden orientar el pronóstico el tipo de la glomerulonefritis y la enfermedad sistémica subyacente. Los casos de positividad simultánea para anticuerpos antimembrana basal glomerular y ANCA constituyen un subgrupo de peor pronóstico con respecto a la respuesta al tratamiento, la recuperación de la función renal y la mortalidad.[6]
Hay casos de resolución espontánea, sobre todo los asociados con infecciones y proliferación endocapilar, pero siempre persiste algún grado de lesión glomerular que con el tiempo, a través de un mecanismo de hiperfiltración glomerular, puede evolucionar hacia la aparición de proteinuria e insuficiencia renal, relacionada con hipertensión arterial o no. Las recaídas no son infrecuentes y suelen asociarse con un descenso rápido de la pauta inmunosupresora. Un importante predictor de la evolución para todos los tipos de glomerulonefritis con formación de semilunas es la gravedad de la insuficiencia renal en el momento de iniciar el tratamiento.[22] Los pacientes que se presentan con insuficiencia renal dependiente de diálisis y cien por ciento de semilunas[23] en general no abandonan los métodos dialíticos a pesar de la administración del régimen inmunosupresor recomendado por diferentes autores.[17]
En vista de la baja probabilidad de recuperación de la función renal en estos casos se ha planteado que los riesgos de la inmunosupresión sobrepasan el beneficio potencial. Sin embargo, en pacientes jóvenes con evidencias de semilunas celulares en la biopsia y que aún no están anúricos se podría considerar la aplicación de una terapia agresiva.[6][8]
Véase también
Notas
- Algunos autores incluyen otros dos tipos en la clasificación: el tipo IV, que representa una combinación del tipo I (anticuerpos antimembrana basal glomerular) con el tipo III (asociado con ANCA), y el tipo V, en el que las características de las lesiones son similares a las del tipo III pero no se detectan ANCA. Muchos identifican esta forma como la verdadera glomerulonefritis semilunar.[4]
Referencias
- MedlinePlus (Enciclopedia médica en español), “Glomerulonefritis rápidamente progresiva”, actualizado en agosto de 2007. Disponible en (enlace roto disponible en Internet Archive; véase el historial, la primera versión y la última).. Consultado el 3 de mayo de 2015.
- Kumar V., Abbas A.K., Fausto N., Robbins S.L. y Cotran R.S., Robbins and Cotran Pathologic Basis of Disease, 7a ed., St. Louis, MO, Elsevier Saunders, 2005, pp. 976-978, ISBN 0-7216-0187-1.
- Kumar V., Cotran R.S. y Robbins S.L., Patología humana, 8a ed., Elsevier Health Sciences, 2008. ISBN 8480868406, 9788480868402. Vista previa disponible en. Consultado el 3 de junio de 2015.
- Roca Goderich R., “Glomerulonefritis rápidamente progresiva”, en Temas de Medicina Interna, t. 2, 2a parte, “Enfermedades del riñón”, cap. 8, “Enfermedades glomerulares”, Ecimed, La Habana, Cuba, 2002. Consultado en el 5 de junio de 2015.
- Rosenberg H., Nefropatías primarias”, en Lecciones de anatomía patológica, consultado en Archivado el 8 de mayo de 2015 en Wayback Machine. el 6 de junio de 2015.
- Sociedad Española de Nefrología, “Glomerulonefritis rápidamente progresiva. Caso 3”. Consultado en el 4 de junio de 2015.
- Lodish H. et al., Biología celular y molecular, Editorial Médica Panamericana, 2005, 973 pp. ISBN 9500613743, 9789500613743. Vista previa disponible en . Consultado el 3 de junio de 2015.
- Pusey C.D., “Antiglomerular basement membrane disease. Kidney Int 2003;64:1535-1550.
- Salama A.D., Levy J.B., “Tolerance and autoimmunity in Anti-GBM Disease”, J Am Soc Nephrol 2003;14:2988-2989.
- Kluth D.C., Rees A.J., “Anti-Glomerular basement membrane disease”, J Am Soc Nephrol 1999;10:2446-2453.
- Rodríguez-Iturbe B., “Glomerulonefritis rápidamente progresiva”, Medwave 2003 Mar;3(2):e774 doi: 10.5867/medwave.2003.02.774. Consultado en el 6 de junio de 2015.
- Manual Merck, “Síndrome nefrítico rápidamente progresivo”, en “Trastornos del riñón y de las vías urinarias” (sección 11). Consultado en el 6 de junio de 2015.
- Rees A.J. y Cameron J.S., “Crescentic glomerulonephritis", en Cameron J.S., Davidson A.M. et al., Oxford Textbook of Clinical Nephrology, Oxford: Oxford University Press,1998: 626-6446.
- Nitta K., “Serum vascular endothelial growth factor concentration in rapidly progressive glomerulonephritis”, Nephron 1998; 80(3):357-358.
- Jennette J.C., “Rapidly progressive crescentic glomerulonephritis”, Kidney Int. 2003, 63(3):1164-1177.
- Salama A.D., Dougan T., Levy J.B., Cook H.T., Morgan S.H., Naudeer S. et al., “Goodpasture's disease in the absence of circulating anti–glomerular basement membrane antibodies as detected by standard techniques”, Am J Kidney Dis 2002, 39(6):1162-1167.
- Benítez Llanes O., Gómez Barry H., Magrans Buch Ch. y Benítez García M. del C., “Insuficiencia renal rápidamente progresiva por enfermedad antimembrana basal glomerular”. Consultado en Archivado el 11 de septiembre de 2012 en Wayback Machine. el 1 de junio de 2015.
- Cole E., Cattran D., Magil A., Greenwool C. et al., A prospective randomized trial of plasma exchange as additive therapy in idiopathic crescentic glomerulonephritis", Am J Kidney Dis 1992;20(3):261-269.
- Rodríguez P., Lorenzo I., Incháustegui L. y Rodríguez M.L., "Neumonía por Pneumocystis carinii en pacientes no trasplantados sometidos a tratamiento prolongado con esteroides. Importancia de la profilaxis con cotrimoxazol", Nefrología 1998;18(1):105-106.
- Furuta T., Hotta O., Yusa N., Horigome I et al., “Lymphocytapheresis to treat rapidly progressive glomerulonephritis: a randomised comparison with steroid-pulse treatment”, Lancet 1998; 352 :203-204.
- Lee R., D’Cruz D., "Novel Therapies for Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis". Disponible en .
- Savage C.O.S., “ANCA–associated renal vasculitis”, Kidney Int 2001;60: 1614-1627.
- Levy J.B., Turner A.N., Rees A.J. y Pusey C.D, “Long–term outcome of anti–glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression”, Ann Intern Med 2001;134(11):1033-1042.
Enlaces externos
 Wikimedia Commons alberga una categoría multimedia sobre Nefrología.
Wikimedia Commons alberga una categoría multimedia sobre Nefrología.
 Datos: Q1509367
Datos: Q1509367- Multimedia: Rapidly progressive glomerulonephritis / Q1509367