Hemofilia
La hemofilia es un trastorno hemorrágico hereditario en el cual la sangre no se coagula de manera adecuada. Esto puede causar sangrados tanto espontáneos como después de una operación o de tener una lesión. Está relacionada con el cromosoma X en los dos principales tipos: la hemofilia A, cuando hay un déficit del factor VIII de coagulación y la hemofilia B, cuando hay un déficit del factor IX de coagulación. Casi en todos los casos esta enfermedad es recesiva para las mujeres, pues si alguno de los padres es hemofílico el otro cromosoma heredado vendrá de un padre sano. Este no es el caso de los hijos varones pues en ellos existe una alta probabilidad de heredar el cromosoma X enfermo.
| Hemofilia | ||
|---|---|---|
| Especialidad | hematología | |
| Síntomas | Dolor en articulaciones, articulación inflamada, hematomas, hemorragia interna, hemorragia nasal, períodos intensos o prolongados, sangrado en heces y/o en la orina | |
| Causas | Usualmente genética. | |
Historia
Antigüedad
Los estudios más antiguos datan desde el siglo II d. C., cuando rabinos judíos se dieron cuenta de que algunos niños varones, cuando se les practicaba la circuncisión, sangraban mucho. Los rabinos descubrieron que estos problemas solo ocurrían en ciertas familias, por lo que hicieron nuevos reglamentos para ayudar a estos niños que sangraban. El rabino Judá declaró que un niño que tuviese hermanos mayores con problemas de sangrado no tenía que ser circuncidado y el rabino Simeón ben Gamaliel impidió que un niño fuese circuncidado porque los hijos de las tres hermanas de la madre se habían desangrado hasta morir.
Entre las referencias escritas posteriores merece destacar la descripción de la enfermedad que hizo en el siglo XI un médico árabe de Córdoba, llamado Abulcasis.
En el siglo XII, otro rabino llamado Maimónides descubrió que, si los niños tenían hemofilia, eran las madres las que la transmitían. Entonces hizo una ley nueva: si una madre tenía hijos con este problema de sangrado y ella se volvía a casar, ninguno de sus nuevos descendientes varones deberían ser circuncisos.
La primera referencia en la Europa cristiana se da en Italia, en 1525, por Alejandro Benedicto.
Edad Contemporánea
En 1800, un médico estadounidense llamado John Conrad Otto (1774-1844) realizó su primer estudio sobre familias hemofílicas, y en el año 1803 descubrió la genética de la hemofilia A. Encontró que madres sin problema de sangrado podían transmitir hemofilia a sus hijos, y sus hijas podían transmitir a sus nietos y bisnietos.
En 1928, el Dr. Hopff describe la enfermedad por primera vez con la palabra «hemofilia».
La historia del tratamiento de la hemofilia comienza en 1900, que se realizaron transfusiones de sangre fresca para aumentar la longevidad de las personas con hemofilia ―dado que el promedio de vida era aproximadamente de 13 años―.
- En 1950 se dio inicio de la disposición del plasma para el tratamiento.
- En 1965 se dio a la disposición del crioprecipitado del plasma.
- En 1970 se inició la producción de concentrados por factores de coagulación.
- En 1980, se dio una baja de este tratamiento debido a que el 80 % de los que padecían hemofilia se contagiaron del VIH/sida.
- A raíz de esto, en 1985, cada uno de los factores fueron sometidos a una inactivación viral, haciendo cada vez el proceso más antiséptico.
- En 1992 salió al mercado el primer factor que no fue derivado del plasma, obtenido por tecnología ADN recombinante.
- En 1995 se introdujo el primer tratamiento profiláctico, capaz de reemplazar el factor debilitado.[1]
El caso más famoso de la hemofilia fue el del último zarevich de Rusia, Alekséi Nikoláyevich Románov (1904-1918), pasado por su madre transmisora, Alejandra Fiódorovna Románova (1872-1918), nieta de la reina Victoria del Reino Unido (1819-1901), transmisora también de esta enfermedad. También, destacan los casos de hemofilia en la realeza española en los hijos de Alfonso XIII (1886-1941) y la reina Victoria Eugenia (1887-1969), nieta también de la reina Victoria del Reino Unido, y que afectaron profundamente a la monarquía española.
Clasificación
Hemofilia A
La hemofilia A se produce cuando hay déficit en el factor VIII de coagulación.
Hemofilia B
Cuando hay un déficit del factor IX de coagulación. La incidencia de la hemofilia B es de 1 por cada 32000 varones vivos.[2]
Hemofilia B de Leyden
Se trata de una forma variante de la hemofilia B, es menos común. Muestra una característica extremadamente inusual de expresión dependiente de la edad. Los valores del factor IX son inferiores al 1 % durante la infancia con lo que la enfermedad es muy grave en niños, después de la pubertad los valores se elevan hasta alrededor del 50 % de lo normal y el trastorno se vuelve asintomático.
Hemofilia C
Cuando hay un déficit del factor XI de coagulación.
Etiología
En cada célula humana hay 46 cromosomas: la mitad la recibimos como herencia de la madre y la otra mitad del padre. Los cromosomas contienen las instrucciones necesarias para ordenar a las células cómo fabricar las proteínas que el organismo requiere para su funcionamiento. Estas instrucciones se encuentran contenidas en pequeñas formaciones que se llaman genes, constituidos de ADN, que son la estructura básica de la vida.
Los cromosomas vienen en pares, por lo que tenemos dos copias de todos nuestros genes; si hay algún daño en algún gen o un cromosoma, hay una copia de respaldo de ese gen o cromosoma que podrá cumplir las funciones normalmente. Pero hay una excepción, los cromosomas sexuales: X y Y.
El sexo femenino está determinado por dos cromosomas X (XX), y el sexo masculino tiene un cromosoma X y un Y (XY). El cromosoma X contiene muchos genes que son comunes a ambos sexos, como los genes para la producción del factor VIII y el factor IX, relacionados con la coagulación sanguínea.
La mujer tiene dos copias de esos genes específicos mientras que los varones solo uno. Si el varón hereda un cromosoma con un gen dañado del factor VIII, es el único gen que recibe y no tiene información de respaldo, por lo que no podrá producir ese factor de coagulación.
Esta anomalía hereditaria se manifiesta en las mujeres, pero en muy bajo porcentaje, ya que las mujeres normalmente son portadoras del gen, igualmente están expuestas a sus consecuencias, ya que para manifestar la enfermedad necesitarían dos copias defectuosas, cosa muy poco probable. Actualmente, en España, la incidencia de personas nacidas con hemofilia es 1 de cada 15 000. Incluso debido a la inactivación del cromosoma X que todas las mujeres hacen, puede ser que mujeres portadoras del gen defectuoso muestren un genotipo de hemofilia leve.[3]
Síntomas
La característica principal de la hemofilia A y B es la hemartrosis y el sangrado prolongado espontáneo. Las hemorragias más graves son las que se producen en articulaciones, cerebro, ojo, lengua, garganta, riñones, hemorragias digestivas, genitales, nasales, etc.
La manifestación clínica más frecuente en los pacientes con hemofilia es la hemartrosis, sangrado intraarticular que afecta especialmente a las articulaciones de un solo eje como la rodilla, el codo o el tobillo. Si se produce una hemartrosis en repetidas ocasiones en una articulación, se origina una deformidad y atrofia muscular llamada artropatía hemofílica.[4]
Como posibles síntomas se puede clasificar en tres estados: I, II y III; el estado I comprende hormigueo en las articulaciones y fiebre en dicha parte del cuerpo, esta se relaciona por tener una hemorragia con traumatismo. El estado II, este hormigueo pasa a sentirse ya como dolor, aún permanece la fiebre en la parte afectada del cuerpo y se acompaña por inflamación debida al cúmulo de sangre presente en la articulación ósea, debido a una hemorragia espontánea con sangrado severo. Como último estado, el estado III se presenta la articulación viscosa e inflamada debido al debilitamiento muscular, rigidez matinal (todo lo referente a la mañana), dolor crónico y movimiento limitado por la obstrucción en la articulación. Interpretado como hemorragia espontánea en músculo y articulación.[1]
Diagnóstico
El diagnóstico del tipo de hemofilia y su nivel de gravedad se hace mediante la historia clínica y un análisis de sangre para la medición, en el laboratorio, a través de pruebas especiales de coagulación, de los grados de los diferentes factores. El objetivo es establecer la severidad de la enfermedad y decidir el tratamiento más adecuado a seguir por el paciente. Actualmente, apenas se utilizan los valores de los distintos factores para la detección de portadores o el diagnóstico prenatal, pues el análisis de ligamiento o análisis directo de las mutaciones permiten resultados más fiables.
Pruebas de laboratorio
| Condición | Tiempo de protrombina | Tiempo de tromboplastina | Tiempo de sangría | Conteo de plaquetas |
|---|---|---|---|---|
| Hemofilia | Sin alteración | Prolongado | Sin alteración | Sin alteración |
Tratamiento
No hay en la actualidad ningún tratamiento curativo disponible (a excepción de un trasplante hepático), y lo único que se puede hacer es corregir la tendencia hemorrágica administrando por vía intravenosa el factor de coagulación que falta, el factor VIII o el IX.
Como posibles tratamiento existe el casero que consiste en el reposo corporal y sobre la zona afectada, dejando hielo durante dos horas sobre dicha parte del cuerpo y elevando posteriormente la articulación.[1]
El tratamiento sustitutivo supuso un avance importantísimo tanto para la calidad de vida como para la supervivencia de los pacientes. La obtención de factores de coagulación a partir de plasma humano dio lugar, en muchas ocasiones, a la transmisión de virus, sobre todo el virus VIH (causante del sida) en los años ochenta, lo que significó un grave retroceso en la vida de los pacientes con hemofilia.
A mediados de esa misma década se introdujeron los primeros métodos de inactivación viral en los concentrados liofilizados, transformándolos en productos mucho más seguros.
Actualmente, los concentrados liofilizados de doble inactivación viral constituyen derivados del plasma más seguros y se están evaluando e introduciendo constantemente tecnología punta en estos productos que permitan inactivar nuevos virus y otros agentes infecciosos, como los priones, que podrían representar una amenaza para los que utilizan productos derivados del plasma humano.
En los últimos años, el desarrollo de la ingeniería genética ha hecho posible iniciar una nueva era en el tratamiento de la enfermedad. Desde hace unos pocos años, se han desarrollado preparados más puros de los factores de coagulación, sin necesidad de plasma humano. El factor VIII recombinante, el más masificado, se produce a partir de células cultivadas en laboratorio.
En las últimas décadas, el foco de las terapias de hemofilia se ha enfocado en reemplazar el factor de coagulación faltante; sin embargo, la biotecnología combinada con un mayor entendimiento de la bioquímica de la coagulación está actualmente cambiando el paradigma del tratamiento.
Diferentes tipos de agentes hemostáticos se encuentran disponibles para el manejo de la hemofilia A. Estos se dividen en terapias de reemplazo y terapias de no reemplazo. Las terapias de reemplazo mejor conocidos como concentrados de factor, son el tratamiento a elegir para las personas con hemofilia, debido a que son seguros y efectivos para tratar y prevenir sangrados, por ejemplo: factor ocho, factor nueve, productos de vida media extendida, agentes puente o de derivación, factor siete recombinante, concentrado complejo protrombínico. También existen las terapias de no reemplazo que no son factores, como lo es Emicizumab, la primera y única terapia de sustitución hasta la fecha. Los beneficios claves de esta terapia son su vía de administración subcutánea y la reducción en la frecuencia de los episodios de sangrado en pacientes con o sin inhibidores.
La Federación Mundial de Hemofilia recomienda: El uso de concentrados de factor derivados de plasma o recombinantes con inactivación viral, evitando el uso de plasma fresco congelado o crioprecipitado, debido a temas de calidad, seguridad y eficacia. Todos los pacientes con inhibidores del factor ocho exógeno deben ser considerados para profilaxis regular para prevenir sangrados. Los pacientes con hemofilia A e inhibidores del factor ocho exógeno, deben estar en profilaxis con emicizumab.
Como medicamento se utiliza principalmente desmopresina para desprender factor VIII de los vasos sanguíneos,[5] cabe resaltar que no se debe utilizar la aspirina convencional (ácido acetilsalicílico) pues esta es un antiagregante plaquetario, y no dejara hacer un trombo en alguna laceración corporal, esto sería un agravante para el estado del paciente. Y como medida evidente, se encuentra el suministro del factor faltante por vía intravenosa ya sea por medio de reservas como plasma fresco congelado, crioprecipitados, pero como medida recomendada, factor liofilizado del factor a utilizar.[6]
Existen enormes expectativas de tratamiento mediante terapia génica, que consiste en la introducción de genes en células determinadas del paciente que sean capaces de combinarse con el material genético existente, aportando la información que falta para fabricar la proteína deficitaria causante de la enfermedad. La hemofilia es buena candidata para terapia génica ya que no requiere la regulación de los genes de los factores VIII o IX insertados (incluso la sobreexpresión del gen no resultaría contraproducente, ya que los genes tienen de forma natural una gran variabilidad), es fácil acceder a las células para la terapia ex vivo, existen buenos modelos animales, y un pequeño aumento en los niveles plasmáticos sería suficiente para convertir una patología grave en una forma más leve (además los niveles son fácilmente medibles en cualquier hospital).
El objetivo de la fisioterapia en el tratamiento de la hemofilia, como profilaxis, es aconsejar y programar actividades físicas y deportes con riesgos mínimos, que prevengan la aparición de lesiones músculo-esqueléticas consecutivas a una deficiente condición física.
Por otra parte, respecto al tratamiento de lesiones, la fisioterapia muestra su colaboración y su resolución del episodio hemorrágico, actúa sobre la inflamación, disminuye el dolor y recupera la función perdida, procurando evitar o disminuir las secuelas.
Hay dos tipos de tratamiento: Profilaxis (el enfermo se inyecta por vía intravenosa su medicina correspondiente varias veces a la semana), o a demanda (el enfermo se inyecta la medicina cada vez que se produce una hemorragia).
De igual manera, la reunión de médicos especialistas de Buenos Aires,[6] ―en conjunto con la Federación Mundial de Hemofilia―[7] concuerdan en la utilización de la ecuación para el reemplazo del factor VIII (con resultados en UI) que corresponde a:
- peso del paciente × porcentaje faltante del factor VIII × 0,5
Se dice que es mejor manejar la infusión del factor liofilizado por productos farmacéuticos, no se recomienda el uso de criprecipitados, ya que a estos no se les ha hecho un barrido viral, y por el uso de este puedan haber muchas más patologías. Consideran la estabilidad del paciente 8 horas antes de cualquier intervención quirúrgica por lo menos de 8 horas de seguimiento y suministrando el factor faltante o deficiente. Recalca que el uso de Desmopresina es exclusivo para la prevención mas no para un tratamiento quirúrgico. Trazando un límite de diferencia entre los factores, y más específicamente entre la ecuación para la suministración, el factor IX maneja casi la misma ecuación, pero es más específica en el ciclo vital de las personas, ya que es distinto el cálculo para niños que para adultos; para niños el cálculo es:
- peso × porcentaje de factor a infundir × 1,5
En cambio para adultos es:
- peso × porcentaje de factor a infundir × 1,2.
Pronóstico
Hoy en día la supervivencia de un paciente con hemofilia es alta, gracias al suministro por vía intravenosa del factor antihemofílico. Las personas que padecen esta enfermedad pueden llevar una vida completamente normal con un tratamiento adecuado.
Directrices de actuación con pacientes con hemofilia
- Nunca suministrar aspirina (ácido acetilsalicílico) a un paciente con hemofilia.
- Está totalmente contraindicada la inyección intramuscular.
- Está desaconsejado el uso de antiinflamatorios tipo ibuprofeno.
- Cobertura hemostática con factor.
Situaciones en las que necesitan acudir al hospital
Es importante saber cuándo se debe ir al hospital y con quién se puede contactar para consultar acerca de un problema de hemorragia antes de que se presente. Determinadas circunstancias personales o condiciones locales por proximidad a un centro especializado, pueden modificar la posibilidad de obtener asistencia hospitalaria, pero todas estas situaciones son las que precisan una consulta rápida y un tratamiento inmediato:
- Dolor en articulaciones o músculos. No esperar a que la hinchazón sea visible.
- Hemorragia externa que no puede ser detenida o que recurre después de un tratamiento de primeros auxilios.
- Sangre en la orina o en las heces.
- Después de una caída con golpe en la cabeza u otra lesión en la cabeza, o si existe dolor de cabeza o náuseas y vómitos prolongados sin causa justificada.
- Hemorragia o hinchazón en la zona alrededor del cuello.
- Dolor abdominal inexplicable.
- Es importante a la hora de llegar a un centro hospitalario, decir la condición de hemofilia y preguntar por el hematólogo del centro.
Herencia
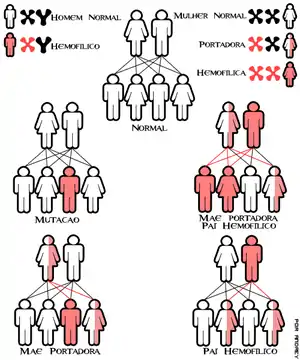
La hemofilia A y la hemofilia B son de herencia gonosómica (sexual, ligada al cromosoma X); el gen alterado en la hemofilia A se localiza en el locus , y el de la hemofilia B, en .


Así pues, tiene una prevalencia mucho mayor en los varones, donde actúa como carácter holándrico:
- Con un padre con hemofilia y madre sana no portadora: el 100 % de sus hijas serán portadoras sanas (heredan el alelo mutado del padre), y el 100 % de los hijos serán sanos no portadores (no tienen de quién recibir el X mutado).
- Con un padre con hemofilia y madre sana portadora (heterocigota): el 50 % de las hijas serán portadoras sanas y el 50 % de las hijas serán hemofílicas. En cuanto a los hijos varones, el 50 % serán pacientes con hemofilia (pues reciben un único X materno, que en este caso es el mutado) y el 50 % serán sanos no portadores (han recibido el X sin defecto).
- Con un padre sano[8] y madre portadora sana: el 50 % de las hijas serán sanas no portadoras, y el 50 % serán sanas portadoras. En cuanto a los hijos varones, al igual que en el caso anterior, el 50 % serán pacientes con hemofilia y el 50 % serán sanos no portadores.
La hemofilia C es de herencia autosómica recesiva, con igual frecuencia en ambos sexos.
La hemofilia es una afección que padecen casi exclusivamente los varones, y casi todos los pacientes con hemofilia son hijos de madres sanas, portadoras del gen, es decir que en sus antepasados existió algún paciente, pero en casi un tercio de los pacientes ha ocurrido sin que haya un historial familiar. En estos casos, la hemofilia es producida por una mutación en el gen de la madre o del niño.
La enfermedad ha sido especialmente estudiada porque afecta a gran parte de las familias reales europeas, por lo menos aquellas cuyos miembros descienden de la reina Victoria de Inglaterra quien transmitió la hemofilia a uno de sus hijos (que falleció de hemorragia interna tras una caída) y a al menos a dos de sus hijas que fueron las que extendieron la enfermedad por las familias reales europeas. No se conocían casos de hemofilia entre los antepasados de la reina Victoria y esto era normal, se supone que la enfermedad apareció por mutación de uno de los alelos normales de esta.
Como vivo ejemplo de la manera en que se propaga esta patología, está el árbol genealógico de la reina Victoria del Reino Unido.
Asesoramiento genético
En las familias en las que algún miembro se encuentre afectado es importante detectar las mujeres con riesgo de ser portadoras y realizar el asesoramiento genético. Lo ideal es realizar este asesoramiento antes de que cualquier mujer con riesgo de la familia se plantee tener descendencia.
El asesoramiento debe considerar dos aspectos: los datos que indican la gravedad de las manifestaciones hemorrágicas y el conocimiento de que las mujeres de una familia con parientes con hemofilia son portadoras de la enfermedad.
La familia debe conocer las implicaciones de la enfermedad, cómo se hereda, la probabilidad de que vuelva a suceder y las alternativas que existen. El asesoramiento genético debe ser un proceso educativo e informativo, pero de ningún modo impositivo.
Véase también
Referencias
- Antonio Liras. La hemofilia. 4 de septiembre de 2010.
- «ESPINOSA, BENJAMÍN GARCÍA; CAMPAL, FAUSTINA RUBIO; GONZÁLEZ, MARÍA ROSARIO CRESPO (1 de enero de 2015). Técnicas de análisis hematológicos. Ediciones Paraninfo, S.A. ISBN 9788428335232.».
- «¿Qué es la hemofilia?», artículo en español en el sitio Portal Hemofilia (España).
- The orthopaedic status of severe haemophiliacs in Spain, páginas 170-176.
- «Hemofilia», artículo en español en el sitio web MedlinePlus (Estados Unidos).
- Consenso de médicos especialistas en hemofilia en la República Argentina: «Guía de tratamiento de la hemofilia» Archivado el 28 de mayo de 2016 en Wayback Machine., artículo publicado en el sitio web Hemofilia.com.ar. Buenos Aires (Argentina), julio de 2011.
- Protocolo para el tratamiento de la hemofilia y de la enfermedad de Von Willebrand.
- Nótese que no cabe la posibilidad de un varón portador.
Enlaces externos
- Federación Mundial de Hemofilia
- SOCHEM. Sociedad Chilena de la Hemofilia
- ASANHEMO. Asociación Andaluza de Hemofilia
- HEMOARALAR. Asociación de hemofilia Aragón-La rioja
- HEMOBUR. Asociación de hemofilia de Burgos
- ASHEMADRID. Asociación de hemofilia de Madrid
- FEDHEMO. Federación Española de Hemofilia
- Real Fundación Victoria Eugenia
- Ashemadrid. Asociación de Hemofilia de la Comunidad de Madrid
- Federación de Hemofilia de la República Mexicana, A. C.
- National Hemophilia Foundation (USA)
- En MedlinePlus hay más información sobre Hemofilia
- OMIM (On-line Mendelian Inheritance in Man), Haemophilia A
- OMIM (On-line Mendelian Inheritance in Man), Haemophilia B
- Hemofilia, F. M. (abril de 2008). Protocolo para el tratamiento de la hemofilia y de la enfermedad de Von Willebrand. Georgia, Estados Unidos
- Grupo DíasMundialesDe (2016-). Día Mundial de la Hemofilia
| Control de autoridades |
|
|---|
 Datos: Q134003
Datos: Q134003 Multimedia: Hemophilia / Q134003
Multimedia: Hemophilia / Q134003