Poroqueratosis
La poroqueratosis (del griego πορος, 'poro' y κɛρατωσις, 'queratosis') es una enfermedad rara de la piel, clasificada dentro de los trastornos de la queratinización epidérmica (CIE-11, ED52: Poroqueratosis).[1][2][3][4][5][6] Se caracteriza por la aparición de una o varias placas hiperqueratósicas de forma redondeada y borde elevado, que tienen un crecimiento centrifugo. Fue descrita por primera vez en 1893 por el dermatólogo italiano Vittorio Mibelli, en un artículo publicado en la revista italiana Giornale italiano di dermatologia e venereologia.[7] El nombre se debe a la creencia errónea por parte de Mibelli de que el trastorno se originaba en las glándulas sudoríparas. No debe confundirse con la paraqueratosis, que es otra entidad dermatológica diferente.[8][9][10]
| Poroqueratosis | ||
|---|---|---|
 Lesión en paciente con poroqueratosis actínica superficial diseminada (PASD) | ||
| Especialidad | Dermatología, Pediatría y Podología | |
| Duración | Enfermedad crónica y progresiva | |
| Causas | En general, desconocidas | |
| Factores de riesgo | Predisposición genética, radiación ultravioleta e inmunodepresión | |
| Diagnóstico | Mediante biopsia de una lesión | |
| Tratamiento | Sin tratamiento específico | |
| Pronóstico | Condición precancerosa | |
| Frecuencia | En general, desconocida | |
| Sinónimos | ||
| ||
Descripción

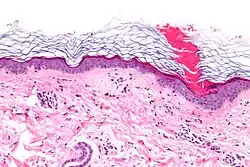
Al estudiar la biopsia de una lesión con ayuda del microscopio, se observa en todos los tipos de poroqueratosis una formación característica llamada laminilla cornoide que está formada por una columna compacta de células hiperqueratósicas en disposición inclinada. Este hallazgo explica la existencia de un borde periférico elevado en las lesiones, cuando se observan a simple vista.
En general la causa de la enfermedad es desconocida, aunque se ha demostrado que en la mayoría de sus formas de presentación existe un factor hereditario autosómico dominante.[11] Se han identificado mutaciones en los genes MVK,[12][13] PMVK,[14][15] MVD,[16][17] FDPS,[18][19] SLC17A9,[1][20][21] SART3[22][23] y se ha observado, por ejemplo, que sólo las mutaciones en el gen de la mevalonato quinasa (MVK) localizado en el cromosoma 12q21, son la causa de hasta un tercio de los casos de poroqueratosis actínica superficial diseminada (POROK4).[24] A veces, diferentes formas de la enfermedad se asocian entre sí y aparecen conjuntamente.[11][25]
Las lesiones de la piel son persistentes y suelen permanecer indefinidamente, en ocasiones con periodos de mejoría y otros de recidiva. Es esencial considerar su potencial malignidad que, aunque no es frecuente, ocurre en algunas ocasiones (alrededor del 10% de los casos)[26] con el desarrollo de un cáncer de piel de tipo carcinoma epidermoide, carcinoma espinocelular, enfermedad de Bowen o carcinoma basocelular.[11] Las variedades más predispuestas serían la poroqueratosis de Mibelli (POROK1) y la poroqueratosis lineal.[27] Las lesiones que tienen un mayor riesgo de degeneración maligna son las de mayor tiempo de evolución, las que se dan en personas mayores, las del tipo lineal y las de gran tamaño en extremidades.[11] Se ha observado también, en pacientes inmunosuprimidos, que la malignización es más precoz e invasiva, con tendencia a metástasis.[27]
La poroqueratosis es una enfermedad crónica y progresiva que puede dar lugar a un impacto negativo en la calidad de vida de los individuos afectados, como consecuencia de la propia presencia de las lesiones en zonas visibles y la necesidad extrema de exponerlas lo menos posible a cualquier tipo de radiación ultravioleta, tanto natural (luz solar) como artificial.[25][26][27] Entre los principales factores de riesgo para el desarrollo de la enfermedad se incluyen la predisposición genética, la exposición a la luz ultravioleta, la inmunodepresión y el tratamiento con algunos fármacos.[24][25][26] Aunque no existe un tratamiento específico, el uso de determinadas terapias permite mejorar ligeramente la calidad de vida del paciente.[11]
Formas de presentación
Existen diversas formas de presentación y cada una de ellas posee unas manifestaciones específicas:
- Poroqueratosis actínica superficial diseminada (ORPHA 79152, OMIM 175900). Es la variante más frecuente. Se manifiesta en la 3.ª y 4.ª década de la vida y sólo en raras ocasiones, durante la infancia. Ocurre más en mujeres que hombres, en una relación 1,8:1. Las lesiones suelen localizarse en áreas fotoexpuestas (frecuentemente en las extremidades) pero nunca en las palmas de las manos o en las plantas de los pies. El 15% de los casos pueden tener afectación facial.[24][27]
- Poroqueratosis clásica de Mibelli (ORPHA 735, OMIM 175800). Es la segunda variante más común y suele manifestarse en la infancia, afectando principalmente a varones en una relación 3:1. Puede aparecer también en la adolescencia o edad adulta, cuando va asociada a individuos inmunosuprimidos. Las lesiones suelen localizarse en las extremidades, pero también pueden afectar los hombros y la región genital. Su presencia en la cara y en las mucosas es poco frecuente.[26][27]
- Poroqueratosis lineal (OMIM 175900 y OMIM 614714). Aparece en recién nacidos y durante la pubertad, predominando en mujeres. Las lesiones se localizan principalmente en las extremidades y pueden afectar a las palmas de las manos y las plantas de los pies.[27]
- Poroqueratosis palmoplantar de Mantoux (ORPHA 736).
- Poroqueratosis palmoplantar diseminada (ORPHA 737, OMIM 175850). Suele aparecer en la adolescencia, aunque en algunos casos se manifiesta en la edad adulta. Por lo general, las lesiones se manifiestan como pápulas queratósicas en las palmas de las manos, plantas de los pies, mucosa oral y lesiones anulares en el resto del cuerpo.[28][27]
- Poroqueratosis punctata (ORPHA 324561, OMIM 175860). Comienza generalmente durante la infancia.[29]
- Craneosinostosis - anomalías anales - poroqueratosis (ORPHA 85199, OMIM 603116). Es un síndrome muy raro (inferior a 1/1.000.000) caracterizado por una craneosinostosis y una hipoplasia clavicular, retraso en el cierre de las fontanelas, anomalías anales, malformaciones genitourinarias y erupción cutánea.[30]
- Poroqueratosis superficial y diseminada eruptiva de Respighi. Suele aparecer en la infancia y en la pubertad. Las lesiones se sitúan principalmente en el tronco.[11]
- Poroqueratosis de forma mínima o de Freund. Se manifiesta en la adolescencia.[11]
- Poroqueratosis postrasplante e inmunosupresión. Se ha observado en pacientes que fueron sometidos a trasplantes de diversos órganos (principalmente riñón) y en aquellos que estuvieron sometidos a inmunosupresores, sobre todo con citostáticos. Además suele encontrarse en pacientes inmunosuprimidos, en asociación con algunas patologías como linfomas de células T o infecciones por VIH.[11]
- Poroqueratosis reticulada. Las lesiones se ubican en abdomen, región suprapúbica, genitales y pliegues, pudiendo cursar con prurito.[11]
- Poroqueratosis minuta digitada. Presenta una morfología que semeja proyecciones córneas filiformes, múltiples y asintomáticas, con un borde de 2 a 3 mm de altura que cuando se supera, pueden quebrarse.[11]
- Poroqueratosis eruptiva papulosa y pruriginosa. Su morfología presenta una forma eruptiva y diseminada acompañada de pápulas eritematosas y pruriginosas que desaparecen después de varios meses, dejando lesiones anulares ligeramente pigmentadas.[11]
- Poroqueratosis liquenoide estriada.[31]
Véase también
Referencias
- OMIM: Porokeratosis 1 (POROK1).
- OMIM: Porokeratosis 3 (POROK3).
- OMIM: Porokeratosis 7 (POROK7).
- «Las enfermedades raras: un desafío para Europa». Comisión Europea.
- Romaní de Gabriel, Jorge. Trastornos de la queratinización epidérmica.
- CIE-11, ED52: Poroqueratosis.
- Mibelli, Vittorio (1893). «Contributo alla studio della ipercheratosi dei canali sudoriferi (porokeratosi)». Giornale italiano di dermatologia e venereologia (Torino) 28: 313-355.
- Paraqueratosis. Cínica Universitaria de Navarra. Consultado el 18 de enero de 2019
- Diccionario Enciclopédico Ilustrado de Medicina Dorland. Consultado el 18 de enero de 2019.
- Kumar, Vinay; Fausto, Nelson; Abbas, Abul (2010) Robbins & Cotran Pathologic Basis of Disease (8th ed.). Saunders. Page 1170. ISBN 978-1-4160-3121-5.
- Rodríguez Peralto, J.L.; Garrido, M.; Guerra, A. Poroqueratosis. Archivado desde el original el 3 de septiembre de 2013. Consultado el 27 de noviembre de 2017.
- OMIM: Mevalonate Kinase; MVK.
- NCBI: MVK mevalonate kinase.
- OMIM: Phosphomevalonate Kinase; PMVK.
- NCBI: PMKV phosphomevalonate kinase.
- OMIM: Mevalonate Pyrophosphate Decarboxylase; MVD.
- NCBI: MVD mevalonate diphosphate decarboxylase.
- OMIM: Farnesyl Diphosphate Synthase; FDPS.
- NCBI: FDPS farnesyl diphosphate synthase.
- OMIM: Solute Carrier Family 17 (Vesicular Nucleotide Transporter), Member 9; SLC17A9.
- NCBI: SLC17A9 solute carrier family 17 member 9.
- OMIM: Squamous Cell Carcinoma Antigen Recognized By T Cells 3; SART3.
- NCBI: SART3 spliceosome associated factor 3, U4/U6 recycling protein.
- Orphanet: Poroqueratosis actínica superficial diseminada (ORPHA 79152).
- Medscape: Porokeratosis.
- Orphanet: Poroqueratosis de Mibelli (ORPHA 735).
- Academia Española de Dermatología y Venereología: Poroqueratosis.
- Orphanet: Poroqueratosis palmoplantar diseminada (ORPHA 737).
- Orphanet: Poroqueratosis punctata (ORPHA 324561).
- Orphanet: Craneosinostosis - anomalías anales - poroqueratosis (ORPHA 85199).
- Poroqueratosis liquenoide estriada.
Enlaces externos
- Base de datos de enfermedades raras en Orphanet.
- Base de datos OMIM (Online Mendelian Inheritance in Man) Catálogo McKusick.
- Lista de enfermedades raras de Orphanet en español, por orden alfabético y formato PDF.
 Wikimedia Commons alberga una categoría multimedia sobre Poroqueratosis.
Wikimedia Commons alberga una categoría multimedia sobre Poroqueratosis.
 Datos: Q7230264
Datos: Q7230264- Multimedia: Porokeratosis / Q7230264