Reacción Tsuji–Trost
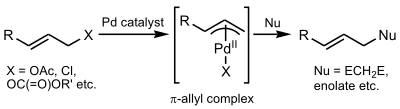
La reacción Tsuji–Trost (también llamada alquilación alílica de Trost o alquilación alílica) es un paladio-catalizador para reacción de sustitución que implica un sustrato que contiene un grupo grupo saliente en una posición alílica. El catalizador de paladio se coordina primero con el grupo alilo y luego sufre una adición oxidativa, formando el complejo π-alilo. Este complejo alílico puede ser atacado por un nucleófilo, dando como resultado el producto sustituido.[1]
Este trabajo fue iniciado por primera vez por Jiro Tsuji en 1965[2] y, posteriormente, fue adaptado por Barry Trost en 1973 con la introducción de ligandos de fosfina.[3] El alcance de esta reacción se ha expandido a muchos nucleófilos a base de carbono, nitrógeno y oxígeno diferentes, muchos grupos salientes diferentes, muchos ligandos a base de fósforo, nitrógeno y azufre diferentes, y muchos metales diferentes (aunque el paladio todavía está privilegiado).[4] La introducción de ligandos de fosfina condujo a una reactividad mejorada y numerosas estrategias de alquilación alílica asimétrica. Muchas de estas estrategias son impulsadas por el advenimiento de los ligandos quirales, que a menudo son capaces de proporcionar una alta Enantioselectividad y alta diastereoselectividad en condiciones suaves. Esta modificación amplía enormemente la utilidad de esta reacción para muchas aplicaciones sintéticas diferentes. La capacidad de formar enlaces carbono-carbono, carbono-nitrógeno y carbono-oxígeno en estas condiciones, hace que esta reacción sea muy atractiva para los campos de la química médica y la síntesis de productos naturales.
Historia
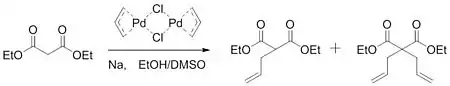
En 1962, Smidt publicó un trabajo sobre la oxidación catalizada por paladio de alquenos a grupos carbonilo. En este trabajo, se determinó que el catalizador de paladio activaba el alqueno para el ataque nucleofílico del hidróxido.[5] Al obtener una idea de este trabajo, Tsuji planteó la hipótesis de que podría tener lugar una activación similar para formar enlaces carbono-carbono. En 1965, Tsuji informó sobre un trabajo que confirmó su hipótesis. Al hacer reaccionar un dímero de cloruro de alilpaladio con la sal sódica de malonato de dietilo, el grupo pudo formar una mezcla de producto monoalquilado y dialquilado.[6]
El alcance de la reacción se expandió gradualmente hasta que Trost descubrió el siguiente gran avance en 1973. Al intentar sintetizar homólogos de sesquiterpeno acíclicos, Trost tuvo problemas con el procedimiento inicial y no pudo alquilar sus sustratos. Estos problemas se superaron con la adición de trifenilfosfina a la mezcla de reacción.
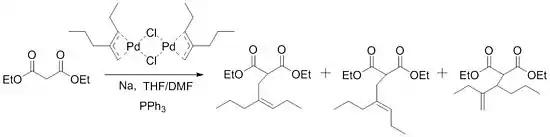
Estas condiciones luego se probaron para otros sustratos y algunos condujeron a una "reacción esencialmente instantánea a temperatura ambiente". Poco después, desarrolló una forma de usar estos ligandos para la síntesis asimétrica.[7] No es sorprendente que esto haya estimulado muchas otras investigaciones de esta reacción y haya llevado al importante papel que esta reacción tiene ahora en la química sintética.
Mecanismo
Empezando con una especie de paladio de valor cero y un sustrato que contiene un grupo saliente en la posición alílica, la reacción de Tsuji-Trost se desarrolla a través del ciclo catalítico descrito a continuación.

Primero, el paladio se coordina con el alqueno, formando un complejo η2 π-alilo- Pd0 Π. El siguiente paso es la adición oxidativa en la que el grupo saliente se expulsa con la inversión de la configuración y se crea un η3 π-alilo- PdII (también llamado ionización). El nucleófilo luego se agrega al grupo alilo que regenera el complejo η2 π-alilo-Pd0. Al finalizar la reacción, el paladio se separa del alqueno y puede comenzar nuevamente en el ciclo catalítico.[8]
Nucleófilos "Duros" contra "blandos"
Los nucleófilos utilizados se generan típicamente a partir de precursores (pronucleófilos) in situ después de su desprotonación con base.[9] Estos nucleófilos se subdividen en nucleófilos "duros" y "blandos" utilizando un paradigma para describir los nucleófilos que se basa en gran medida en las pKas de sus ácidos conjugados. Los nucleófilos "duros" típicamente tienen ácidos conjugados con pKas mayores que 25, mientras que los nucleófilos "duros" típicamente tienen ácidos conjugados con pKas menores que 25.[10] Este descriptor es importante debido al impacto que estos nucleófilos tienen en la estereoselectividad del producto. Estabilizado o "suave"del complejo π-alilo. Esta inversión junto con la inversión en estereoquímica asociada con la adición oxidativa de paladio produce una retención neta de estereoquímica. Los nucleófilos no estabilizados o "duros", por otro lado, retienen la estereoquímica del complejo π-alilo, lo que resulta en una inversión neta de la estereoquímica.
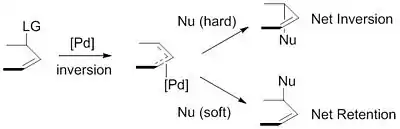
Esta tendencia se explica al examinar los mecanismos de ataque nucleofílico. Los nucleófilos "suaves" atacan el carbono del grupo alilo, mientras que los nucleófilos "duros" atacan el centro metálico, seguido de la eliminación reductiva.
Ligandos de Fosina
Los ligandos de fosfina, como la trifenilfosfina o el ligando de Trost, se han utilizado para ampliar considerablemente el alcance de la reacción de Tsuji-Trost. Estos ligandos pueden modular las propiedades del catalizador de paladio, como el volumen estérico y las propiedades electrónicas. Es importante destacar que estos ligandos también pueden inculcar quiralidad en el producto final, lo que hace posible que estas reacciones se lleven a cabo de forma asimétrica, como se muestra a continuación.
Sustitución asimétrica alílica
La versión enantioselectiva de la reacción de Tsuji-Trost se denomina alquilación alílica asimétrica de Trost (Trost AAA) o simplemente, alquilación alílica asimétrica (AAA). Estas reacciones se utilizan a menudo en síntesis asimétrica.[11][12][13] La reacción se desarrolló originalmente con un catalizador de paladio soportado por el ligando de Trost, aunque las condiciones adecuadas se han expandido mucho desde entonces. La enantioselectividad puede impartirse a la reacción durante cualquiera de los pasos aparte de la descomplejación del paladio del alqueno desde el estereocentro.Ya está establecido en ese punto. Se han conceptualizado cinco formas principales para aprovechar estos pasos y generar condiciones de reacción enantioselectivas. Estos métodos de enantiodiscriminación fueron revisados previamente por Trost:
- Ionización preferencial a través de la complejación de olefinas enantioselectivas
- Ionización Enantiotópica De Grupos Salientes
- Ataque a Términos enantiotopicos del Complejo Allyl
- Intercamabio de enantioface en el complejo π-Allilo
- Diferenciación de Prochiral Nucleophile Caras
El método favorecido para enantiodiscriminación es en gran parte dependiente en el substrato de interés, y en algunos casos, el enantioselectividad puede ser influido por varios de estos factores.
Alcance
Nucleófilos
Se ha informado que muchos nucleófilos diferentes son efectivos para esta reacción. Algunos de los nucleófilos más comunes incluyen malonatos, enolatos, alcóxidos primarios, carboxilatos, fenóxidos, aminas, azidas, sulfonamidas, imidas y sulfonas.
Saliendo de grupos
El alcance de los grupos salientes también se ha ampliado para incluir varios grupos salientes diferentes, aunque los más utilizados son los carbonatos, fenoles, fosfatos, haluros y carboxilatos.
Nucleófilos "duros" y "blandos"
Trabajos recientes han demostrado que el alcance de los nucleófilos "blandos" se puede expandir para incluir algunos pronucleófilos que tienen pKas mucho más altos que ~ 25. Algunos de estos nucleófilos "blandos" tienen pKas que van hasta 32,[14] e incluso más Se ha demostrado que los pronucleófilos básicos (~ 44) actúan como nucleófilos blandos con la adición de ácidos de Lewis que ayudan a facilitar la desprotonación.[15] La gama pKa mejorada de nucleófilos "blandos" es crítica porque estos nucleófilos son los únicos que han sido exploradosref>Trost, B. M.; Toste, F. D. J. Am. Chem. Soc. 1999, 121, 4545.</ref>[16] para reacciones enantioselectivas hasta hace muy poco tiempo[17] (aunque reacciones no enantioselectivas de "duras"). Al aumentar el alcance de los pronucleófilos que actúan como nucleófilos "blandos", estos sustratos también pueden incorporarse en reacciones enantioselectivas utilizando métodos previamente descritos y bien caracterizados.
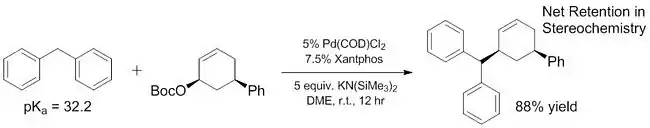
Ligandos
Sobre la base de la reactividad del ligando de trifenilfosfina, la estructura de los ligandos utilizados para la reacción de Tsuji-Trost se volvió rápidamente más compleja. Hoy en día, estos ligandos pueden contener fósforo, azufre, nitrógeno o alguna combinación de estos elementos, pero la mayoría de los estudios se han concentrado en los ligandos de mono y difosfina. Estos ligandos pueden clasificarse adicionalmente según la naturaleza de su quiralidad, con algunos ligandos que contienen quiralidad central en los átomos de fósforo o carbono, algunos con quiralidad biaril axial y otros con quiralidad planar. Los ligandos de difosfina con quiralidad central surgieron como un tipo eficaz de ligando (particularmente para los procedimientos de alquilación alílica asimétrica) con el ligando de Trost como uno de esos ejemplos.[18] Se han empleado ligandos de fosfinooxazolinas (PHOX) en el AAA, particularmente con nucleófilos basados en carbono.[19]
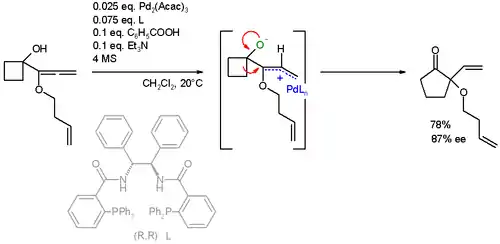
Aplicaciones
Síntesis/de productos farmacéuticos / naturales
La capacidad de formar enlaces carbono-carbono, carbono-nitrógeno y carbono-oxígeno enantioselectivamente en condiciones suaves hace que la alquilación alílica asimétrica de Trost sea extremadamente atractiva para la síntesis de moléculas complejas.
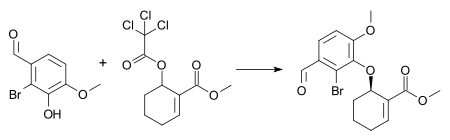
Un ejemplo de esta reacción es la síntesis de un intermedio en la síntesis total combinada de galantamina y morfina[22] con 1% en moles [dímero de cloruro de pi-alilpaladio], 3% en moles (S, S) ligando de Trost y trietilamina en diclorometano a temperatura ambiente. Estas condiciones dan como resultado la formación del enantiómero (-) del éter arílico con un rendimiento químico del 72 % y un exceso enantiomérico del 88 %.
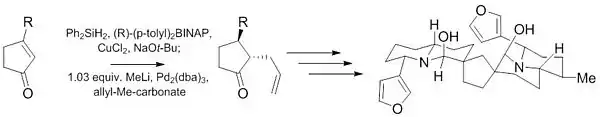
Se utilizó otra reacción de Tsuji-Trost durante las etapas iniciales de la síntesis de (-) - Neothiobinupharidine. Este trabajo reciente demuestra la capacidad de esta reacción para proporcionar productos altamente diastereoselectivos (10: 1) y enantioselectivos (97.5: 2.5) a partir de material de inicio aquiral con solo una pequeña cantidad de catalizador (1%).[23]
Detección de paladio
Aparte de la aplicación práctica de esta reacción en la química médica y la síntesis de productos naturales, trabajos recientes también han utilizado la reacción de Tsuji-Trost para detectar paladio en varios sistemas. Este sistema de detección se basa en un no fluorescente de fluoresceína sensor derivada de (sensores de longitud de onda más largas tienen también ha sido recientemente desarrollado para otras aplicaciones[24]) que se vuelve fluorescente sólo en la presencia de paladio o platino. Esta capacidad de detección de paladio / platino es impulsada por la reacción Tsuji-Trost. El sensor contiene un grupo alilo con la fluoresceína funcionando como el grupo saliente. El complejo π-alilo se forma y después de un ataque de nucleófilos, se libera la fluoresceína, lo que produce un aumento dramático en la fluorescencia.[25][26]
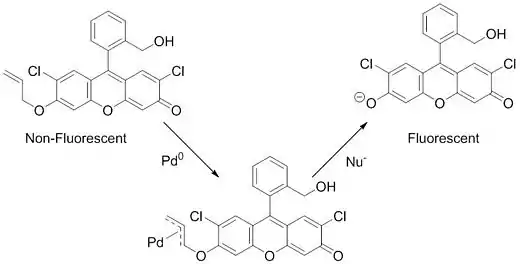
Este simple, método de alto rendimiento para detectar paladio mediante el control de la fluorescencia se ha demostrado ser útil en el seguimiento de los niveles de paladio en minerales metálicos,[27] productos farmacéuticos,[28] e incluso en las células vivas.[29] Con la creciente popularidad de la catálisis de paladio, este tipo de detección rápida debería ser muy útil para reducir la contaminación de los productos farmacéuticos y prevenir la contaminación del medio ambiente con paladio y platino.
Enlaces externos
- Org. Synth. 1989, 67, 105
- Org. Synth. 2009, 86, 47
- Ejemplo de tsuji-trost la reacción en síntesis total ve : http://www.biocis.u-psud.fr/img/pdf/concise_total_synthesis_of_minfiensine.pdf la segunda reacción encontrada encima sitio web del biocis equipo : http://www.biocis.u-psud.fr/spip.php?article332
Referencias
- Strategic Applications of Named Reactions in Organic Synthesis (Paperback) by Laszlo Kurti, Barbara Czako
- Organic syntheses by means of noble metal compounds XVII. Reaction of π-allylpalladium chloride with nucleophiles Tetrahedron Letters, Volume 6, Issue 49, 1965, Pages 4387-4388 Jiro Tsuji, Hidetaka Takahashi, Masanobu Morikawa doi 10.1016/S0040-4039(00)71674-1
- Trost, B. M.; Fullerton, T. J. "New synthetic reactions. Allylic alkylation." J. Am. Chem. Soc. 1973, 95, 292–294. doi 10.1021/ja00782a080.
- Rios, Itzel Guerrero; Rosas-Hernandez, Alonso; Martin, Erika; "Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation." Molecules, 2011, 16 970–1010. doi 10.3390/molecules16010970
- Smidt, J., Hafner, W., Jira, R., Sieber, R., Sedlmeier, J. and Sabel, A. (1962), Olefinoxydation mit Palladiumchlorid-Katalysatoren. Angewandte Chemie, 74: 93–102. doi 10.1002/ange.19620740302
- Organic syntheses by means of noble metal compounds XVII. Reaction of Plantilla:Pi-allylpalladium chloride with nucleophiles Tetrahedron Letters, Volume 6, Issue 49, 1965, pages 4387–4388 Jiro Tsuji, Hidetaka Takahashi, Masanobu Morikawa doi 10.1016/S0040-4039(00)71674-1
- Asymmetric Transition Metal-Catalyzed Allylic Alkylations Barry M. Trost David L. Van Vranken Chem. Rev., 1996, 96 (1), pp 395–422 doi 10.1021/cr9409804
- Trost, Barry M.; Zhang, Ting; Sieber, Joshua D.; "Catalytic asymmetric allylic alkylation employing heteroatom nucleophiles: a powerful method for C-X bond formation." Chem. Sci. 2010, 1, 427–440.
- Strategic Applications of Named Reactions in Organic Synthesis (Paperback) by Laszlo Kurti, Barbara Czako ISBN 0-12-429785-4
- Trost, B. M.; Thaisrivongs, D. A. J. Am. Chem. Soc. 2008, 130, 14092
- Trost, B. M.; Dietsch, T. J. "New synthetic reactions. Asymmetric induction in allylic alkylations." J. Am. Chem. Soc. 1973, 95, 8200–8201. doi 10.1021/ja00805a056.
- Trost, B. M.; Strege, P. E. "Asymmetric induction in catalytic allylic alkylation." J. Am. Chem. Soc. 1977, 99, 1649–1651. doi 10.1021/ja00447a064.
- Asymmetric Transition-Metal-Catalyzed Allylic Alkylations:Applications in Total Synthesis Trost, B. M.; Crawley, M. L. Chem. Rev.; (Review); 2003; 103(8); 2921–2944. doi 10.1021/cr020027w
- Sha, Sheng-Chun; Zhang, Jiadi; Carroll, Patrick J.; Walsh, Patrick J.; "Raising the pKa Limit of "Soft" Nucleophiles in Palladium-Catalyzed Allylic Substitutions: Application of Diarylmethane Pronucleophiles." JACS. 2013, 135, 17602–17609. doi: 10.1021/ja409511n
- Zhang, J.; Stanciu, C.; Wang, B.; Hussain, M. M.; Da, C.-S.; Carroll, P. J.; Dreher, S. D.; Walsh, P. J. Palladium-Catalyzed Allylic Substitution with (η6-Arene–CH2Z)Cr(CO)3-Based Nucleophiles, J. Am. Chem. Soc. 2011, 133, 20552.
- Trost, B. M.; Machacek, M. R.; Aponick, A. Acc. Chem. Res. 2006, 39, 747.
- Li, Xiao-Hui; Zheng, Bao-Hui; Ding, Chang-Hua; Hou, Xue-Long; "Enantioselective Synthesis of 2,3-Disubstituted Indanones via Pd-Catalyzed Intramolecular Asymmetric Allylic Alkylation of Ketones." Org. Lett. ASAP. doi: 10.1021/ol402980v
- Lu, Zhan; Ma, Shengming; "Metal-Catalyzed Enantioselective Allylation in Asymmetric Synthesis." Angew. Chem. Int. Ed. 2008, 47, 258–297. doi: 10.1002/anie.200605113
- Behenna, D. C.; Stoltz, B. M., Shengming; "The Enantioselective Tsuji Allylation." J. Am. Chem. Soc. 2004, 126, 15044–15045. doi: 10.1021/ja044812x
- Trost, B. M.; Xie, J. "Palladium-Catalyzed Asymmetric Ring Expansion of Allenylcyclobutanols: An Asymmetric Wagner–Meerwein Shift." J. Am. Chem. Soc. 2006, 128, 6044–6045. doi 10.1021/ja0602501.
- The co-catalysts are benzoic acid and triethylamine. Molecular sieves (MS) prevent hydrolysis.
- Trost, B. M.; Tang, W.; Toste, F. D. "Divergent Enantioselective Synthesis of (−)-Galantamine and (−)-Morphine." J. Am. Chem. Soc. 2005, 127, 14785–14803. doi 10.1021/ja054449+.
- Jansen, Daniel J.; Shenvi, Ryan A.; "Synthesis of (−)-Neothiobinupharidine." JACS. 2013, 135, 1209–1212. doi: 10.1021/ja310778t
- Wang, Zhifei; Zheng, Shuang; Cai, Jin; Wang, Peng; Feng, Jie; Yang, Xia; Zhang, Liming; Ji, Min; Wu, Fugen; He, Nongyue; Wan, Neng; "Fluorescent Artificial Enzyme-Linked Immunoassay System Based on Pd/C Nanocatalyst and Fluorescent Chemodosimeter." Anal. Chem. ASAP. doi: 10.1021/ac403001y
- Garner, Amanda L.; Koide, Kazunori; "Studies of a fluorogenic probe for palladium and platinum leading to a palladium-specific detection method." Chem. Commun. 2009, 86–88. doi:10.1039/b814197e
- Song, Fengling; Garner, Amanda L.; Koide, Kazunori; "A Highly Sensitive Fluorescent Sensor for Palladium Based on the Allylic Oxidative Insertion Mechanism." JACS. 2007, 129, 12354–12355. doi: 10.1021/ja073910q
- Williams, Jessica M.; Koide, Kazunori; "A High-Throughput Method To Detect Palladium in Ores." Ind. Eng. Chem. Res. 2013, 52, 8612–8615. doi: 10.1021/ie400959z
- Bu, Xiaodong; Koide, Kazunori; Carder, Evan J.; Welch, Christopher J.; "Rapid Analysis of Residual Palladium in Pharmaceutical Development Using a Catalysis-Based Fluorometric Method." Org. Process Res. Dev. 2013, 17, 108–113. doi: 10.1021/op3003008
- Zhu, Baocun; Gao, Chenchen; Zhao, Yunzhou; Liu, Caiyun; Li, Yamin; Wei, Qin; Ma, Zhenmin; Du, Bin; Zhang, Xiaoling; "A 4-hydroxynaphthalimide-derived ratiometric fluorescent chemodosimeter for imaging palladium in living cells." Chem. Commun. 2011, 47, 8656–8658. doi: 10.1039/c1cc13215f
| Control de autoridades |
|
|---|
 Datos: Q900629
Datos: Q900629 Multimedia: Tsuji-Trost reaction / Q900629
Multimedia: Tsuji-Trost reaction / Q900629