Síndrome de Williams
El síndrome de Williams o síndrome de Williams-Beuren, también llamado monosomía 7, es un trastorno genético poco común causado por una pérdida de material genético en el cromosoma 7, descrito por primera vez en 1961 por el cardiólogo neozelandés John Williams y paralelamente por el pediatra alemán Alois Beuren.
| Síndrome de Williams | ||
|---|---|---|
| Especialidad |
genética médica pediatría | |
| Síntomas | Cambios faciales que incluyen estructura de mentón subdesarrollada, discapacidad intelectual, baja estatura | |
| Complicaciones | Problemas cardíacos , períodos de niveles altos de calcio en la sangre. | |
| Duración | Duración de por vida | |
| Diagnóstico diferencial | Síndrome de Noonan , síndrome de alcoholismo fetal , síndrome de DiGeorge [1] | |
| Sinónimos | ||
| Síndrome de Williams-Beuren | ||
Historia
Este síndrome fue descrito por primera vez en el año 1961 por el Dr. J.C.P. Williams, médico cardiólogo neozelandés que informó de un cuadro clínico complejo, cuyos síntomas más destacados consistían en un retraso general en el desarrollo mental, una expresión característica de la cara y un defecto de nacimiento, conocido como estenosis supravalvular aórtica (ESA) y consistente en un estrechamiento de la aorta en las proximidades del corazón. Paralelamente, el Profesor Beuren, especialista en pediatría de la ciudad alemana de Gotinga, informó de varios casos de ESA, que presentaban una sintomatología similar a la descrita por el Dr. Williams. Posteriormente, en el año 1964, el Profesor Beuren demostró que en estos cuadros clínicos también aparecen frecuentemente estrechamientos de las arterias pulmonares (pulmonalestenosis periférica o SP).
El cuadro descrito por ambos científicos es conocido en Europa a veces como síndrome de Beuren o síndrome de Williams-Beuren, aunque cada vez más se conoce simplemente como síndrome de Williams.
Epidemiología
Es un trastorno de origen genético que se presenta, según estimaciones, en uno de cada 20 000 nacimientos vivos. Afecta igualmente a hombres y mujeres y no tiene preferencia étnica.
Etiología
La causa del Síndrome de Williams es una pérdida de parte del material genético en la banda 7q11.23. de uno de los dos cromosomas 7 del ADN, procedente del padre o de la madre. Se trata de una pérdida submicroscópica, lo que significa que no se aprecia bien cuando se visualiza al microscopio.
La alteración tiene su origen antes de la formación del embrión, bien en el óvulo o bien en el espermatozoide, tras haber sufrido una pérdida de genes en el cromosoma 7 durante su formación mediante división celular o meiosis. Por ello, hasta donde se sabe, no es hereditario.[2][3]
El número de genes perdidos todavía no ha sido determinado, pero se estima que oscila entre los 20 y 30, de los 80.000 que existen. La pérdida de esos genes puede causar que las funciones que dirigen no se realicen normalmente. Sin embargo, no todas las funciones de los genes ausentes son anómalas dado que existe otro cromosoma completo en el par 7.
Queda mucho por conocer sobre el sustrato genético del síndrome de Williams, no obstante se sabe que uno de los genes ausentes es el que produce la elastina, una proteína que da elasticidad a los vasos sanguíneos y otros tejidos corporales. La pérdida de este gen es nociva, porque parece que es necesario tener ambas copias del mismo para la producción de elastina en cantidades adecuadas. La reducción en el suministro de elastina podía ser la responsable de varias patologías derivadas del Síndrome de Williams, como la Estenosis Supravalvular Aórtica (ESVA) y las hernias, así como de la aparición prematura de arrugas. Sin embargo las alteraciones cognitivas o del comportamiento derivan de la ausencia de otros genes, como el WSTF[4] y el FKBP6,[4] responsables de la codificación de proteínas activas en el cerebro que podrían influir en el desarrollo y en las funciones del mismo.
Cuadro clínico
Los síntomas del síndrome son un conjunto de patologías médicas específicas, trastornos psicológicos y signos externos, que se manifiestan durante el desarrollo del individuo, por lo general no antes de los 2 o 3 años de vida del mismo, y que no siempre confluyen todos juntos en la misma persona. También se suelen presentar síntomas parecidos a los de la depresión: falta de interés por las cosas y soledad.
Patologías
Son frecuentes los problemas cardiovasculares, tales como la estenosis aórtica supravalvular y la hipercalcemia transitoria.
Desarrollo mental
Suele presentarse algún tipo de discapacidad intelectual.
Desarrollo del lenguaje
Dificultades en el desarrollo del lenguaje expresivo y en la comprensión. El lenguaje se adquiere tarde respecto a la edad cronológica de referencia para la adquisición de ciertos hitos y no exento de dificultades de comprensión y articulación.
Conducta
Puede presentarse un comportamiento inusualmente alegre y tranquilo ante los desconocidos, unido a impredecibles arrebatos de mal humor o malestar.
Percepción emocional
Facilidad en comprender el estado mental de sus interlocutores (empatía). Este aspecto ha sido puesto en relación con el autismo. Sin embargo las personas con el síndrome de Williams por lo general poseen habilidades sociales muy buenas. De hecho Temple Grandin, autora de Pensar con imágenes: mi vida con el autismo, ha afirmado que las anomalías en el cerebro de quienes padecen este síndrome son contrarias a las del autismo.
Percepción espacial
Inhabilidad en la visualización de cómo diferentes partes pueden unirse con el fin de crear objetos mayores, por ejemplo la unión de las piezas de un rompecabezas.
Un equipo de investigadores del Instituto Nacional de la Salud Mental utilizó imágenes de resonancia magnética nuclear para observar el flujo sanguíneo del cerebro en varios individuos sometidos a dos tareas que implicaban relaciones espaciales. Las personas con síndrome de Williams mostraron menor actividad en la sección del cerebro asociada a las relaciones espaciales que las personas sin síndrome. Esto indica un déficit de dispersión en el tejido en el sistema visual del cerebro, que percibe las relaciones espaciales. Este déficit obstruye parte de la transmisión de información visual.
Otro experimento documentó que las personas con síndrome de Williams, cuando se les muestra una imagen, dibujan los detalles pequeños pero no el conjunto. De esta manera se puede generalizar que los pacientes con síndrome de Williams "pueden ver los árboles, pero no el bosque".
Zurdismo
Tendencia al zurdismo y al uso del ojo izquierdo.
Musicalidad
Las personas que tienen el síndrome suelen tener pasión por la música y en ellos son más frecuentes los casos de oído absoluto.
Fisonomía
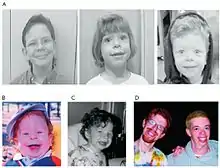
Las personas con el síndrome suelen tener una apariencia facial denominada élfica, alargamiento de las facciones, caballete nasal bajo y una muy acentuada distancia entre la nariz y la boca.
Enfermedades asociadas
- Oftalmológicas:
- Vasculares:
- Soplo cardiaco;
- Estenosis de diversas índoles:
- Estenosis aórtica supravalvular;
- Estenosis aórtica;
- Estenosis de las arterias pulmonares o supraaórtica;
- Estenosis múltiple en arterias pulmonares periféricas;
- Estenosis pulmonar;
- Estenosis de la arteria renal.
- Defecto septal ventricular (DSV);
- Defecto septal auricular (ADS);[5]
- Hipertensión.
- Renales
- Problemas en la sangre
- Digestivas
- Estreñimiento crónico.
Diagnóstico
El retraso madurativo, la presencia de un trastorno circulatorio, el fracaso escolar o el aspecto facial, son algunos de los indicadores del síndrome, que al ser detectados por los pediatras, cardiólogos infantiles o neurólogos, hacen que se derive al paciente al genetista, que lo diagnostica.
Actualmente es posible confirmar el diagnóstico por métodos moleculares en más del 95% de los casos. El criterio que se sigue para el diagnóstico molecular del Síndrome de Williams es la detección de la deleción de la llamada región crítica del Síndrome de Williams Beuren (WBCSR) que abarca al gen de la elastina (ELN). El 99% de los individuos con diagnóstico clínico de Síndrome de Williams presentan dicha deleción, que puede ser detectada usando técnicas de FISH o análisis de mutación específica.
FISH
El método diagnóstico más utilizado, aunque no el único posible, se denomina FISH (Hibridación In Situ Fluorescente). Esta prueba consiste en aplicar un reactivo sobre la región del cromosoma 7q11.23, de un trozo de ADN obtenido de una célula del individuo, normalmente de la sangre. La fluorescencia aparece cuando algún gen de la pareja de cromosomas 7 no está duplicado.
Análisis de mutación específica
Dentro de este grupo se engloban diversos métodos no basados en FISH utilizados para la detección de deleciones en la región WBSCR:
PCR cuantitativa a tiempo real. La PCR cuantitativa a tiempo real es utilizada para determinar la dosis (el número de copias) de tres genes que se encuentran en la región WBSCR: ELN, LIMK y GTF2I. El hallazgo de una sola copia de una región génica indica la presencia de una deleción de la región WBSCR.
Análisis por microarray El uso de arrays comercialmente disponibles basados en la hibridación genómica comparativa detecta cambios en el número de copias en la deleción de la región WBSCR.
Prueba de heterocigosis Se testan pequeñas repeticiones en tandem (STRs) que abarcan a la región WBSCR. El hallazgo de dos tamaños diferentes de STR (heterocigosidad) en todos los marcadores indica que no hay ninguna deleción presente. Por el contrario, el hallazgo de un único tamaño de STR para todos los marcadores puede indicar homocigosis para todos los marcadores o la presencia de una posible deleción. Se puede llevar a cabo una PCR cuantitativa para determinar si hay realmente una deleción. El análisis de STR se realiza fundamentalmente para determinar el tamaño de las deleciones.
Un diagnóstico certero y precoz es fundamental para evitar pasos innecesarios y planificar las medidas óptimas de seguimiento y tratamiento.
Tratamiento
Al ser un trastorno genético no existe un tratamiento de curación para el síndrome de Williams, sino que se tratan las alteraciones de salud, del desarrollo y de la conducta que se presentan en cada caso particular.
Cada uno de esos trastornos debe ser correctamente atendido por el especialista que corresponda: estimulador temprano, psicomotricistas, neuropsicólogo, terapeuta físico, fonoaudiólogo, psicólogo, psicopedagogo, musicoterapeuta, etc. Lo más importante es un diagnóstico temprano que permita de inmediato el adecuado apoyo terapéutico precoz, a través de programas de educación especial individualizados.
Mediante la terapia del desarrollo, la terapia del lenguaje[7] y terapia ocupacional, el objetivo real y alcanzable es la integración social y laboral de estas personas cuando lleguen a ser adultos.
Precauciones
- Los tratamientos deben ser coordinados por un genetista con experiencia en el síndrome de Williams.
- Se debe tener en cuenta que, cada vez que una persona con Síndrome de Williams requiera sedación o anestesia general para alguna cirugía o procedimiento de diagnóstico, se debe realizar una completa evaluación previa, ya que la literatura médica refiere varios casos de efectos adversos.
- En caso de existir niveles altos de calcio en la sangre, deben evitarse los suplementos de calcio y de vitamina D.[8]
- Los niños con Síndrome de Williams, al llegar a la edad escolar se recomienda que ingresen a una escuela de educación normal, pues tienden a la imitación y esto es favorable para ellos por la deficiencia en su intelecto.
- Son niños muy sociables y como padres se debe ser muy cuidadosos, pues tienden a socializar frecuentemente con personas mayores, no importando si son desconocidas, su inhibición es mínima y por lo tanto es preferible ser precavidos.
Referencias
- Morris, Colleen A. (1993). Adam, Margaret P., ed. Williams Syndrome. University of Washington, Seattle. Consultado el 19 de mayo de 2023.
- Al tratarse de una alteración en la división celular que origina el óvulo o espermatozoide, el riesgo de tener un niño con el Síndrome de Williams es siempre el mismo. Por otro lado, hasta la fecha solamente uno o dos individuos con el síndrome han tenido descendencia, por lo que no es posible establecer conclusiones respecto a la transmisión genética del mismo de padres con Síndrome de Williams.
- Si uno de los progenitores tiene Síndrome de Williams, el porcentaje de pasarlo a sus descendientes es de un 50% de manera autosómica dominante. Revista de la Asociación de Síndrome de Williams de España. Número 12. Enero 2013, en http://es.calameo.com/read/002052335eb77fe7f7460. Otros Artículos de interés: Patil SJ1, Salian, Bhat, Girisha, Shrivastava, Vs, Sapare. Familial 7q11.23 duplication with variable phenotype. Am J Med Genet A. 2015 Nov;167(11):2727-30. doi: 10.1002/ajmg.a.37226.
- Lu, Xiaojun; Meng, Xun; Morris, Colleen A.; Keating, Mark T. (1 de diciembre de 1998). «A Novel Human Gene,WSTF,Is Deleted in Williams Syndrome». Genomics (en inglés) 54 (2): 241-249. ISSN 0888-7543. doi:10.1006/geno.1998.5578. Consultado el 19 de abril de 2022.
- Tanto el DSV como el ADS son la división del ventrículo o de la aurícula cardiaca por un tabique.
- La nefrocalcinosis es una insuficiencia renal, debida a la precipitación de fosfato cálcico en los tubos renales.
- La fortaleza verbal de las personas con Síndrome de Williams puede ayudar a compensar otras debilidades derivadas del mismo.
- Pober BR. N Engl J Med 2010; 362:239-252. Review article.
Enlaces externos
- Enciclopedia médica en español - Síndrome de Williams
- The Williams Syndrome Association
- Williams Syndrome Foundation
- Asociación Síndrome de Williams de Andalucía
- Asociación Síndrome de Williams de Ávila
- Asociación Síndrome de Williams España (ASWE)
| Control de autoridades |
|
|---|
 Datos: Q558077
Datos: Q558077 Multimedia: Williams syndrome / Q558077
Multimedia: Williams syndrome / Q558077