Chronoampérométrie
La chronoampérométrie est une technique électrochimique dans laquelle le potentiel de l'électrode de travail est échelonné et le courant résultant des processus faradiques se produisant à l'électrode (provoqué par l'étape potentielle) est surveillé en fonction du temps. La relation fonctionnelle entre le courant et le temps est mesurée après l'application d'un saut de potentiel simple ou double à l'électrode de travail du système électrochimique. Des informations limitées sur l'identité des espèces électrolysées peuvent être obtenues à partir du rapport entre le courant de réduction de point et celui d'oxydation de pointe. Cependant, comme avec toutes les techniques pulsées, la chronoampérométrie génère des courants de charge élevés, qui décroissent exponentiellement avec le temps comme n'importe quel circuit RC. Le courant faradique - qui est dû à des événements de transfert d'électrons et est le plus souvent la composante actuelle d'intérêt - se désintègre comme décrit dans l'équation de Cottrell. Dans la plupart des cellules électrochimiques, cette désintégration est beaucoup plus lente que les cellules de désintégration de charge sans électrolyte de support sont des exceptions notables. Le plus souvent, un système à trois électrodes est utilisé. Le courant étant intégré sur des intervalles de temps relativement plus longs, la chronoampérométrie donne un meilleur rapport signal / bruit par rapport à d'autres techniques ampérométriques.[1],[2],[3].
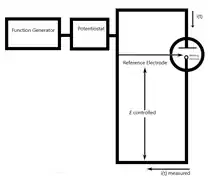
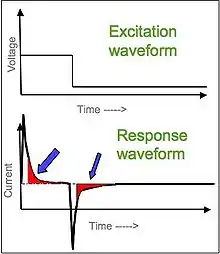
Il existe deux types de chronoampérométrie couramment utilisés, la chronoampérométrie à potentiel contrôlé et la chronoampérométrie à courant contrôlé. Avant d'exécuter la chronoampérométrie à potentiel contrôlé, des voltamètres cycliques sont exécutés pour déterminer le potentiel de réduction des analytes. Généralement, la chronoampérométrie utilise des électrodes à zone fixe, ce qui convient pour étudier les processus d'électrode des réactions chimiques couplées, en particulier le mécanisme de réaction de l'électrochimie organique[4].
Exemple
L'anthracène dans le diméthylformamide désoxygéné (DMF) sera réduit (An + e − → An − ) à la surface de l'électrode qui est à un certain potentiel négatif. La réduction sera limitée à la diffusion, provoquant ainsi une baisse du courant dans le temps (proportionnelle au gradient de diffusion formé par la diffusion).
Vous pouvez faire cette expérience plusieurs fois en augmentant les potentiels d'électrode de bas en haut. (Entre les expériences, la solution doit être agitée.) Lorsque vous mesurez le courant i (t) à un certain point temporel fixe τ après l'application de la tension, vous verrez qu'à un certain moment le courant i (τ) ne monte plus; vous avez atteint la région à transfert de masse limité. Cela signifie que l'anthracène arrive aussi vite que la diffusion peut l'amener à l'électrode.
Histoire
En 1902, FG Cottrell déduit la diffusion linéaire sur une électrode plane selon la loi de diffusion et la transformation de Laplace, et obtient l'équation de Cottrell: , où i est le courant en A, n est le nombre d'électrons, F est la constante de Faraday, A est l'aire de l'électrode plane en cm 2, C 0 est la concentration initiale de l'analyte en mol / cm 3 . D est le coefficient de diffusion des espèces en cm 2 / s, t est le temps en s. Dans des circonstances de diffusion contrôlée, le tracé courant-temps reflète le gradient de concentration de la solution près de la surface de l'électrode. Le courant est directement proportionnel à la concentration à la surface de l'électrode.

En 1922, Jaroslav Herovsky a réitéré la méthode chronoampérométrique lorsqu'il a inventé la méthode polarographique. Il peut utiliser le circuit de base du polarographe. Pour connecter l'enregistreur rapide ou l'oscilloscope, l'électrode de chute de mercure n'est pas utilisée, mais des électrodes statiques telles que le mercure en suspension, le pollen au mercure ou le platine, l'or et le graphite sont utilisés. De plus, la solution n'est pas agitée. En présence d'électrolytes inertes, le processus de transfert de masse est principalement la diffusion[5]. Jarroslav Herovsky a dérivé la méthode chronopotentiométrique de l'équation de Cottrell. La chronopotentiométrie est une méthode électrochimique qui peut générer un courant stable qui peut circuler entre deux électrodes différentes. [6]
Application
Électrolyse (en masse) à potentiel contrôlé
L'une des applications de la chronoampérométrie est l'électrolyse (en masse) à potentiel contrôlé, également connue sous le nom de coulométrie potentiostatique. Au cours de ce processus, un potentiel constant est appliqué à l'électrode de travail et le courant est contrôlé dans le temps. L'analyte dans un état d'oxydation sera oxydé ou réduit à un autre état d'oxydation. Le courant diminuera jusqu'à la ligne de base (s'approchant de zéro) à mesure que l'analyte est consommé. Ce processus montre la charge totale (en coulomb) qui s'écoule dans la réaction. La charge totale (valeur n) est calculée par l'intégration de la zone sous la parcelle actuelle et l'application de la loi de Faraday.
La cellule d'électrolyse (en masse) à potentiel contrôlé est généralement une cellule à deux compartiments (divisée), contenant une anode auxiliaire en tige de carbone et est séparée du compartiment de la cathode par une bougie d'électrolyte à solvant de verre fritté et de méthylcellulose[7]. La raison de la cellule à deux compartiments est de séparer la réaction cathodique et anodique. L'électrode de travail pour l'électrolyse en masse pourrait être un disque RVC, qui a une plus grande surface pour augmenter la vitesse de la réaction[8].

L'électrolyse à potentiel contrôlé est normalement utilisée avec la voltampérométrie cyclique. La voltampérométrie cyclique est capable d'analyser le comportement électrochimique de l'analyte ou de la réaction. Par exemple, la voltampérométrie cyclique pourrait nous indiquer le potentiel cathodique d'un analyte. Puisque le potentiel cathodique de cet analyte est obtenu, l'électrolyse à potentiel contrôlé pourrait maintenir ce potentiel constant pour que la réaction se produise[9],[10].
Chronoampérométrie à double potentiel (DPSCA)
Le DPSCA est une technique dans laquelle l'électrode de travail est appliquée avec un potentiel augmentant pendant un certain temps et diminuant pendant un certain temps. Le courant est surveillé et tracé en fonction du temps. Cette méthode commence par une période d'induction. Au cours de cette période, plusieurs conditions initiales seront appliquées à la cellule électrochimique afin que la cellule puisse s'équilibrer dans ces conditions. [11] Le potentiel de l'électrode de travail sera maintenu au potentiel initial dans ces conditions pendant une période spécifiée (c'est-à-dire généralement trois secondes). Lorsque la période d'induction est terminée, les cellules de travail changent de potentiel pendant un certain temps. Une fois la première étape terminée, le potentiel de l'électrode de travail diminue, généralement au potentiel avant l'étape précédente[12],[13]. L'expérience se termine par une période de relaxation. Au cours de cette période, la condition par défaut implique de maintenir le potentiel de l'électrode de travail de l'état initial pendant encore une seconde environ[14],[15]. Lorsque la période de relaxation est terminée, les conditions de repos post-expérience seront appliquées à la cellule afin que l'instrument puisse revenir à l'état de repos 1. Après avoir tracé le courant en fonction du temps, un chronoampérogramme se produira et il peut également être utilisé pour générer des tracés de Cottrell[16].

Autres méthodes de chrono-analyse
Chronopotentiométrie
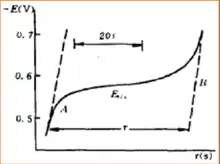
L'application de la chronopotentiométrie pourrait être dérivée en deux parties. En tant que méthode analytique, la plage d'analyse se situe normalement dans la plage de 10 -4 mol / L à 10 -2 mol / L, et parfois elle sera aussi précise que 10 −5 mol / L. Lorsque l'analyse se situe dans la plage de concentration extrêmement faible, une densité de courant inférieure peut être utilisée. De plus, pour obtenir la détermination précise de la concentration, le temps de transition pourrait être prolongé. Dans ce domaine de détermination de l'analyse, la chronopotentiométrie est similaire à la polarographie. Les ondes séparables en polarographie sont également séparables en chronopotentiométrie.
La chronopotentiométrie est une méthode efficace pour étudier le mécanisme de l'électrode. Différentes électrodes auront une relation différente entre E et t dans le graphique de chronopotentiométrie. Dans cette situation, E est le potentiel d'électrode en tension et t est le temps de réaction en secondes. Par la méthode d'étude de la relation entre E et t dans le graphique de chronopotentiométrie, nous pouvons obtenir les informations d'un grand nombre de mécanismes de réactions d'électrodes, telles que la réaction d'électrodes de peroxyde d'hydrogène et d'acide oxalique. L'expérience de la chronopotentiométrie peut se faire dans un laps de temps très court, c'est donc une méthode efficace pour étudier le comportement d'adsorption à la surface de l'électrode. En étudiant le graphe de chronopotentiométrie de l'électrode après adsorption des ions fer, l'existence de l'absorption de platine sur les ions fer est prouvée. En étudiant le graphique de chronopotentiométrie de l'électrode de platine adsorbant l'iode, il est prouvé que l'adsorption de l'iode se produit sous forme de molécules d'iode, et non d'atome d'iode.
Chronocoulométrie
La chronocoulométrie est une méthode analytique d'un principe similaire à celui de la chronoampérométrie, mais elle suit la relation entre la charge et le temps au lieu du courant et du temps. La chronocoulométrie présente les différences suivantes avec la chronoampérométrie : le signal augmente avec le temps au lieu de diminuer ; l'acte d'intégration minimise le bruit, résultant en une courbe de réponse hyperbolique lisse ; et les contributions des charges à double couche et des espèces absorbées sont facilement observables.
Voir également
Références
- Peter Kissinger et William R. Heineman, Laboratory Techniques in Electroanalytical Chemistry, Second Edition, Revised and Expanded, CRC, (ISBN 978-0-8247-9445-3)
- Allen J. Bard et Larry R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, (ISBN 978-0-471-04372-0)
- Cynthia G. Zoski, Handbook of Electrochemistry, Elsevier Science, (ISBN 978-0-444-51958-0)
- J.M. Seveant, « Potential-sweep chronoamperometry: Kinetic currents for first-order chemical reaction parallel to electron-transfer process (catalytic currents) », Electrochimica Acta, vol. 10, no 9, , p. 905–920 (DOI 10.1016/0013-4686(65)80003-2)
- « The Nobel Prize in Chemistry 1959 »
- Peter James Lingane & Dennis G. Peters (1971) Chronopotentiometry, C R C Critical Reviews in Analytical Chemistry, 1:4, 587-634, DOI: 10.1080/1040834nu08542742
- Vanalabhpatana et Peters, « Catalytic Reduction of 1,6-Dihalohexanes by Nickel(I) Salen Electrogenerated at Glassy Carbon Cathodes in Dimethylformamide », J. Electrochem. Soc., vol. 1152, no 7, , E222–E229 (DOI 10.1149/1.1928168)
- Cleary, Mubarak, Vieira et Anderson, « Electrochemical reduction of alkyl halides at vitreous carbon cathodes in dimethylformamide », Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, vol. 198, no 1, , p. 107–124 (DOI 10.1016/0022-0728(86)90030-6)
- Foley, Du, Griffith et Karty, « Electrochemistry of substituted salen complexes of nickel(II): Nickel(I)-catalyzed reduction of alkyl and acetylenic halides », Journal of Electroanalytical Chemistry, vol. 647, no 2, , p. 194–203 (DOI 10.1016/j.jelechem.2010.06.001)
- Vieira et Peters, « Voltammetric behavior of tertiary butyl bromide at mercury electrodes in dimethylformamide », Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, vol. 196, no 1, , p. 93–104 (DOI 10.1016/0022-0728(85)85083-X)
- Faulkner, L. R.; Bard, A. J. Basic Potential Step Methods, Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New Jersey, 2000; 156-225.
- Cottrell, F. G. Z. Physik, Chem., 42, 1902, 385.
- Kambara, T. Bull. Chem. Soc. Jpn., 1954, 27, 523.
- Hyk, W; Nowicka, A.; Stojek, Z. Anal. Chem., 2002, 74, pp 149–157
- Long, J. W.; Terrill, R. H.; Williams, M. E.; Murray, R. W. Anal. Chem., 1997, 69, pp 5082–5086.
- Schwarz, W. M.; Shain, I. J. Phys. Chem., 1965, 69, pp 30-40.
 Portail de l’électricité et de l’électronique
Portail de l’électricité et de l’électronique