Effets non linéaires en catalyse asymétrique
En catalyse asymétrique (également appelée catalyse énantiosélective), un effet non linéaire fait référence à un processus dans lequel l'énantiopureté du catalyseur (ou de l'auxiliaire chiral) n'est pas proportionnelle à l'énantiopureté du produit obtenu[1],[2].
Une réaction asymétrique catalytique peut être représentée suivant l'équation chimique suivante :
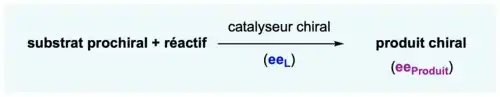
Un substrat prochiral va réagir avec un réactif en présence d'un catalyseur chiral caractérisé par son énantiopureté (ou excès énantiomérique eeL). Le produit de la réaction sera ainsi chiral avec une certaine énantiopureté (ou excès énantiomérique eeProduit). Par exemple, on s'attendrait à ce qu'un catalyseur énantiopur (de 100 % d'excès énantiomérique) générant un produit avec 90 % d'excès énantiomérique devrait générer ce même produit avec un ee de 45 % si le catalyseur avait un ee de 50 %.
La linéarité peut être exprimée mathématiquement avec l'équation :
- .

Pour une réaction asymétrique idéale, le eeProduit peut être décrit comme le produit de ee0 multiplié par le eeL (ee0 est la valeur de l'excès énantiomérique du produit obtenue avec le catalyseur énantiopur). Donc eeproduit = 0,9 × 0,5 = 0,45, soit 45 % ee.
« Ce ne sera pas le cas pour les réactions présentant des effets non linéaires. »[3] Une énantiosélectivité supérieure (ou inférieure) à la valeur proportionnelle de l'excès énantiomérique du catalyseur est considérée comme un comportement non usuel. Cet écart par rapport à la linéarité est appelé « effet non linéaire » ou « NLE ».
Les phénomènes non linéaires seront observés dans des réactions avec une composition de catalyseur ou de réactif scalémique. Cela a été observé pour la première fois par Wynberg et Feringa en 1976 en synthèse asymétrique[4]. Les concepts et les premiers exemples expérimentaux des effets non linéaires en catalyse asymétrique ont été ensuite rapportés et rationalisés par Henri B. Kagan au milieu des années 1980[5]. Les définitions générales et les modèles mathématiques sont essentiels pour comprendre ces effets et leur implication dans des réactions chimiques énantiosélectives. Au cours des dernières décennies, l'étude des effets non linéaires a permis d'élucider le mécanisme de diverses réactions et de guider les applications synthétiques.
Types d'effets non linéaires

Effet non linéaire positif, (+)-NLE
Un effet non linéaire positif, (+)-NLE, est présent dans une réaction asymétrique qui démontre un ee du produit (eeProduit) plus élevé que prévu par rapport à une situation linéaire idéale (cas A au lieu du cas B, figure 1). Il s'agira d'une amplification asymétrique. Un exemple d'effet non linéaire positif est observé dans le cas de l'époxydation de Sharpless avec le substrat géraniol[5].
Effet non linéaire négatif, (−)-NLE
Un effet non linéaire négatif (ou épuisement asymétrique) est présent lorsque le eeproduit est inférieur à celui prédit par une situation linéaire idéale (cas C au lieu du cas B, figure 1). Contrairement à un (+)-NLE, un (-)-NLE entraîne une vitesse de réaction globale plus rapide et une diminution de l'énantiosélectivité. Un exemple intéressant d'un effet (-)-NLE a été rapporté dans les oxydations asymétriques de sulfure.
Modélisation des effets non linéaires
Dès 1986, Henri Kagan et ses collaborateurs ont développé des modèles simples pour expliquer ces phénomènes non linéaires. Tous ces phénomènes ont pour cause des perturbations diastéréomériques[1].
Pour générer un catalyseur énantiosélectif, un métal (symbolisé par la lettre M) sera associé à un ligand (symbolisé par la lettre L) qui sera chiral (LR ou LS). L’association du métal à un ligand générera une espèce de type ML (appelée complexe), l’association du métal à deux ligands donne un complexe de type ML2, puis ML3, ML4, etc. À partir de deux ligands pour un métal, il y a la possibilité de former des diastéréoisomères (exemple pour ML2 : MLRLR, MLSLS et MLRLS). Ces diverses espèces auront une réactivité et énantiosélectivité différentes qui seront à l’origine des effets non linéaires.
En fonction du type de mécanisme, différents modèles sont possibles pour rendre compte de ces effets.
Effet réservoir (modèle simple)
Si le ligand chiral utilisé possède un excès énantiomérique eeL (0 < eeL < 100) pour générer un catalyseur, il est possible qu’une partie du catalyseur soit piégé sous forme d’espèces inactives (le réservoir). Soit α, la fraction de ces complexes non impliqués dans le cycle catalytique. Cette fraction α possédera un excès énantiomérique noté eeres (res pour réservoir). Si α ≠ 0 et eeres ≠ eeL, cela modifiera l’excès énantiomérique du ligand engagé dans le cycle catalytique. Le eeL sera modifié, il donnera un excès énantiomérique efficace noté eeeff. La figure 2 résume la situation.

La relation entre eeeff et eeL sera donnée par l’équation suivante :
- et .


Un exemple de représentation graphique eeeff = f(eeL) est donné sur la figure 3 pour α = 0, α = 0,5 et α = 0,75 en considérant que le réservoir piège une espèce racémique (c’est-à-dire eeres = 0 %). Une partie du ligand sous sa forme racémique étant piégé dans le réservoir, l’excès énantiomérique de l’espèce active (eeeff) sera augmentée par rapport à sa valeur de départ (eeL). Il y aura une amplification de la chiralité qui sera augmentée avec la fraction du réservoir α.

Ce modèle simple ne prenant pas en compte les paramètres cinétiques de la réaction permet de comprendre comment une amplification asymétrique est possible par piégeage d'une partie des espèces catalytiques actives. Le modèle peut être appliqué à des résultats expérimentaux, la figure 4 superpose le modèle « effet réservoir » avec les résultats obtenus pour la réaction d'alkylation de ZnEt2 sur le benzaldéhyde en présence du ligand chiral DAIB[6]. Ce modèle reste cependant très simple et ne peut donner que des droites comme simulation ce qui est éloigné de la réalité expérimentale. Il ne prend pas en compte l'aspect cinétique des réactions et la fraction d'espèce inactive α est constante quelle que soit la valeur de eeL, ce qui ne correspond pas à la réalité des systèmes étudiés.

Modèle MLn
Les modèles communément appelés « MLn » impliquent la présence d’espèces catalytiques qui associent un métal à plusieurs ligands : deux ligands = modèle ML2, trois ligands = modèle ML3, etc.[1]. Dès que l’on associe deux ligands (ou plus) sur un même métal, plusieurs diastéréomères sont possibles. Toutes ces espèces peuvent avoir une concentration et une réactivité différente et donc induire des perturbations. Ces modèles prennent en compte la cinétique de la réaction. Ces modèles s'appliquent également lorsque l'on a des oligomères : dimère (ML)2, trimère (ML)3, etc.
Description
Le cas le plus simple serait l'assemblage de deux ligands chiraux avec un métal pour former un complexe, ce qui donne le modèle ML2. Dans un tel système, trois stéréoisomères sont possibles comme le montre la figure 5. Deux complexes homochiraux (MLRLR et MLSLS, qui donneront les produits avec les excès énantiomériques ee2 et -ee2, respectivement) et un complexe hétérochiral (MLRLS) (souvent appelé « composé méso ») qui donnera le produit racémique (ee = 0 %). Le produit de la catalyse est de ce fait obtenu à partir des trois catalyseurs en équilibre et qui fonctionnent simultanément. Par conséquent, l'excès énantiomérique du produit (eeP) dépendra de (i) l’ee du ligand (eeL), (ii) de la réactivité relative g, avec k2 et k2' étant les constantes cinétiques correspondantes et de (iii) la concentration relative en catalyseur β (avec [RR], [SS] et [RS] définis comme les concentrations individuelles des espèces MLRLR, MLSLS et MLRLS. K est la constante d'équilibre entre les espèces homochirales et hétérochirales.

En prenant en compte tous ces paramètres, il est possible de relier eeP à eeL :

avec .

Interprétation des résultats mathématiques du modèle ML2
- Si β = 0, aucun complexe hétérochiral (MLRLS) n'est présent ; l'équation ML2 se simplifie. Idem si g = 1, les complexes auront la même réactivité. Par conséquent, dans ces deux cas, les propriétés additives simples devraient s'appliquer pour établir une relation linéaire entre l'énantiosélectivité du produit et l'énantiopureté du ligand chiral.
- Si l'on a une distribution statistique des trois espèces chirales, alors la constante d'équilibre sera égale à 4. La figure 6 décrit des simulations de courbes à différentes valeurs de g. Si g = 0 (c'est-à-dire que l'espèce hétérochirale n'est pas active), un effet (+)-NLE est observé. Si g > 1 alors l'espèce hétérochirale est plus active que l'espèce homochirale, un effet (-)-NLE est observé.

- Une situation devient intéressante lorsque la constante K est élevée, autrement dit lorsqu'il y a une prédominance du composé hétérochiral. La figure 7 représente des exemples à différentes valeurs de g pour K = 2 500. Si g = 0 (espèce hétérochirale inactive), l'effet non linéaire positif devient extrêmement important. Cette situation rejoint celle de l'effet réservoir. L'espèce hétérochirale (racémique) joue le rôle de réservoir et induit un fort effet (+)-NLE. Inversement, si g est grand (par exemple g = 100), l'espèce hétérochirale prédomine et comme elle génère le produit sous sa forme racémique, une forte déplétion est observée (effet non linéaire négatif).

Modèle ML3

Description
Sur le même schéma, le modèle peut être étendu à des systèmes plus complexes, par exemple lorsqu'un complexe métallique avec 3 ligands donnant une espèce de type ML3 est formé, ou une espèce trimérique (ML)3. Dans le cas ML3, quatre espèces sont possibles : (MLRLRLR), (MLSLSLS), (MLRLRLS) et (MLSLSLR) (figure 8). Comme dans le modèle ML2, il y a coexistence d'une espèce homochirale et d'une espèce hétérochirale, mais avec une différence majeure : l'espèce hétérochirale n'est pas méso et va générer un produit de réaction qui ne sera pas racémique. Il s'agit de ce fait d'une situation où les deux espèces vont générer le produit avec un certain ee, les courbes des effets non linéaires peuvent être très caractéristiques. La forme devient indicative d'un mécanisme.
Exemples d'effets non linéaires possibles avec le modèle ML3
Un exemple intéressant avec le modèle ML3 est décrit sur la figure 9. Si l'espèce hétérochirale est plus réactive que l'espèce homochirale (grande valeur de g avec g = k3'/k3) et qu'elle induit un produit avec une meilleure énantiosélectivité (c'est-à-dire que ee'3 >> ee3) alors, il serait possible d'avoir un catalyseur plus efficace lorsqu'il n'est pas énantiopure. Si l'on prend les courbes de la figure 9, les paramètres ont été fixés à : ee3 = 50 % ee et ee'3 = 100 % ee, donc l'espèce hétérochirale induit une meilleure énantiosélectivité que l'espèce homochirale. Dès que g est significatif, la courbe montre une zone où le catalyseur est plus efficace sous une forme non énantiopure.
Afin de nommer ce type d'effet non linéaire, Henri Kagan a proposé le terme d'« effet non linéaire hyperpositif »[1]. Le premier exemple expérimental d'effet non linéaire hyperpositif a été observé 26 ans après les hypothèses de Kagan, cependant le mécanisme s'est avéré différent[7]

Modèle ML4
Le modèle ML4 a également été traité par Henri Kagan, il suit le même schéma que les deux modèles ci-dessus. Des modélisations de ce cas ont donné des courbes très caractéristiques.
Exemples expérimentaux d'effets non linéaires et comparaison avec les modèles cinétiques
Époxydation du géraniol (réaction de Sharpless)
Les effets non linéaires de la réaction de Sharpless ont été étudiés avec le géraniol comme substrat par Kagan dès 1986 (figure 10a)[5]. L'effet non linéaire observé est un effet non linéaire positif avec une légère amplification asymétrique comme le montre la figure 10b. En appliquant le modèle de type ML2, il est possible d'avoir une bonne prédiction de la courbe avec les paramètres indiqués sur la figure. Les résultats sont cohérents avec les études menées par Barry Sharpless qui suggéra une espèce dimérique comme espèce active (donc de type (ML)2)[8]. La valeur de g dans la simulation est de 0,35 c'est-à-dire que k'2/k2 ≈ 1/3 ce qui suggère que l'espèce dimère (ML)2 homochirale (figure 10c) est trois fois plus réactive que l'espèce homochirale.

Réaction d'alkylation asymétrique d'aldéhyde
La réaction asymétrique du dialkylzinc et du benzaldéhyde en présence du ligand chiral (-)-3-exo-(diméthylamino)isobornéol (nommé DAIB) présente des phénomènes non linéaires inhabituels ayant fait l'objet de nombreuses études mécanistiques. En présence d'une quantité catalytique de DAIB, la réaction du dialkylzinc et d'un aldéhyde aromatique est nettement accélérée pour donner, après hydrolyse, l'alcool correspondant avec une très grande pureté énantiomérique (figure 11a). L'étude de l'excès énantiomérique du produit en fonction de l'excès énantiomérique du ligand [eeP = f(eeL)] donne un effet non linéaire positif très marqué comme le montre la figure 11b. Par exemple pour R = Et, un excès énantiomérique de 95 % pour le produit est obtenu avec un ligand ayant uniquement 15 % d'excès énantiomérique.

Le mécanisme de la réaction a été étudié par Ryoji Noyori et l'origine de l'effet non linéaire a pu être déterminé[6],[9]. Celui-ci dérive du modèle ML2 de Kagan. Ici, les (+)-NLE proviennent de la formation de dimères homochiraux et hétérochiraux, qui jouent le rôle de réservoir. Contrairement au modèle ML2 de Kagan, la réaction est catalysée par des complexes monomériques tandis que les dimères sont inactifs et en équilibre dynamique avec les monomères. Ici, le NLE est régie purement par les stabilités relatives des espèces dimériques, qui à leur tour modifient le rapport R/S des monomères catalytiquement actifs. Un (+)-NLE se produit lorsque le dimère hétérochiral est plus stable que le dimère homochiral (et inversement). La figure 12 donne un aperçu global du mécanisme.

Références
- D. Guillaneux, S.-H. Zhao, O. Samuel et D. Rainford, « Nonlinear Effects in Asymmetric Catalysis », J. Am. Chem. Soc., vol. 116, , p. 9430–9439 (DOI 10.1021/ja00100a004).
- T. Satyanarayana, S. Abraham et H. B. Kagan, « Nonlinear Effects in Asymmetric Catalysis », Angew. Chem. Int. Ed., vol. 48, , p. 456–494 (PMID 19115268, DOI 10.1002/anie.200705241).
- C. Girard et H. B. Kagan, « Nonlinear Effects in Asymmetric Synthesis and Stereoselective Reactions: Ten Years of Investigation », Angew. Chem. Int. Ed., vol. 37, , p. 2922–2959 (PMID 29711141, DOI 10.1002/(SICI)1521-3773(19981116)37:21<2922::AID-ANIE2922>3.0.CO;2-1).
- H. Wynberg et B. Feringa, « Enantiomeric recognition and interactions », Tetrahedron, vol. 32, , p. 2831–2834 (DOI 10.1016/0040-4020(76)80131-7, lire en ligne).
- (en) C. Puchot, O. Samuel, E. Dunach et S. Zhao, « Nonlinear effects in asymmetric synthesis. Examples in asymmetric oxidations and aldolization reactions », J. Am. Chem. Soc., vol. 108, no 9, , p. 2353–2357 (DOI 10.1021/ja00269a036).
- N. Kitamura, S. Okada, S. Suga et R. Noyori, « Enantioselective Addition of Dialkylzincs to Aldehydes Promoted by Chiral Amino Alcohols. Mechanism and Nonlinear Effect », J. Am. Chem. Soc., vol. 111, , p. 4028-4036 (DOI 10.1021/ja00193a040).
- (en) Y. Geiger, T. Achard, A. Maisse-François et S. Bellemin-Laponnaz, « Hyperpositive nonlinear effects in asymmetric catalysis », Nature Catalysis, vol. 3, no 5, , p. 422–426 (ISSN 2520-1158, DOI 10.1038/s41929-020-0441-1, lire en ligne, consulté le ).
- (en) M. G. Finn et K. Barry Sharpless, « Mechanism of asymmetric epoxidation. 2. Catalyst structure », J. Am. Chem. Soc., vol. 113, no 1, , p. 113–126 (ISSN 0002-7863 et 1520-5126, DOI 10.1021/ja00001a019, lire en ligne, consulté le ).
- M. Kitamura, S. Suga, H. Oka et R. Noyori, « Quantitative Analysis of the Chiral Amplification in the Amino Alcohol-Promoted Asymmetric Alkylation of Aldehydes with Dialkylzincs », J. Am. Chem. Soc., vol. 120, , p. 9800-9809 (DOI 10.1021/ja981740z).
 Portail de la chimie
Portail de la chimie