Complexe de coordination
Un complexe de coordination est constitué d'un atome ou d'ion central, généralement métallique, appelé centre de coordination, et d'un réseau de molécules ou d'ions liés, appelés ligands[1],[2],[3].

De nombreux composés contenant des métaux, en particulier ceux qui comprennent des métaux de transition (éléments tels que le titane qui appartiennent au bloc du tableau périodique), sont des complexes de coordination[4].
Nomenclature et terminologie
Les complexes de coordination sont tellement omniprésents que leur structure et leurs réactions sont décrites de manière variable et confuse. L'atome au sein d'un ligand qui est lié à l'atome ou à l'ion métallique central est dit atome donneur. Dans un complexe typique, un ion métallique est lié à plusieurs atomes donneurs, qui peuvent être identiques ou différents. Un ligand polydenté est une molécule ou un ion qui se lie à l'atome central par l'intermédiaire de plusieurs atomes du ligand. Il est couramment possible de rencontrer des ligands liés à l'atome central avec 2, 3, 4 ou même 6 liaisons. Ces complexes sont appelés chélates ; la formation de tels complexes est appelée chélation, complexation et coordination.
L'atome ou l'ion central, ainsi que tous les ligands, constituent la sphère de coordination[5],[6]. Les atomes ou ions centraux et les atomes donneurs constituent la première sphère de coordination.
La coordination fait référence aux « liaisons covalentes coordonnées » (liaisons dipolaires) entre les ligands et l'atome central. À l'origine, un complexe impliquait une association réversible de molécules, d'atomes ou d'ions par l'intermédiaire de liaisons chimiques faibles. Appliquée à la chimie de coordination, cette signification a évolué. Certains complexes métalliques se forment de manière pratiquement irréversible et beaucoup sont liés entre eux par des liaisons assez fortes[7],[8].
Le nombre d'atomes donneurs liés à l'atome ou à l'ion central est appelé coordinence, ou nombre de coordination. Les nombres de coordination les plus courants sont 2, 4 et surtout 6. Un ion hydraté est un exemple d'ion complexe formé entre un ion métallique central et un ou plusieurs ligands, molécules ou ions environnants qui contiennent au moins un doublet non liant.
Si tous les ligands sont monodentés, alors le nombre d'atomes donneurs est égal au nombre de ligands. Par exemple, l'ion hexahydraté cobalt(II) ou, hexaaquacobalt(II) [Co(H2O)6]2+ est un ion complexe hydraté, en l'occurrence un complexe aqua, composé de six molécules d'eau liées à un cation de cobalt à l'état d'oxydation +2. La denticité des ligands et la coordinence du centre métallique permettent de déduire le nombre de ligands. Ainsi, l'ion complexe bis(éthylènediamine)platine(II), de formule abrégée Pt(en)22+, a une coordinence de 4 et non de 2 car l'éthylènediamine est un ligand bidenté.
Tout atome donneur cède un doublet d'électrons. Certains atomes ou groupes donneurs peuvent céder plus d'un doublet : ils sont dits bidentés (cèdent deux doublet) ou polydentés (cèdent plus de deux doublets). Dans certains cas, un atome ou un groupe cède un doublet d'électrons à deux atomes ou accepteurs métalliques centraux semblables ou différents — par scission du doublet d'électrons — en une liaison à trois centres et deux électrons. On parle alors de ligands pontants.
Histoire

Les complexes de coordination sont connus depuis le début de la chimie moderne. Les premiers complexes de coordination bien connus comprennent des colorants tels que le bleu de Prusse . Leurs propriétés ont été comprises pour la première fois à la fin des années 1800, à la suite des travaux de 1869 de Christian Wilhelm Blomstrand . Blomstrand a développé ce que l'on appelle aujourd'hui la théorie des chaîne. En considérant les complexes d'amines métalliques, il a émis l'hypothèse que les molécules d'ammoniac compensaient la charge de l'ion en formant des chaînes de type [(NH3)X]X+, où X est le nombre de coordination de l'ion métallique. Il a comparé ses chaînes d'ammoniac théoriques à des hydrocarbures de la forme (CH2)X[9].
À la suite de cette théorie, le scientifique danois Sophus Mads Jørgensen y a apporté des améliorations. Dans sa version de la théorie, Jørgensen affirmait que lorsqu'une molécule se dissocie dans une solution, il y aurait deux résultats possibles : les ions se lieraient via les chaînes d'ammoniac décrites par Blomstrand ou les ions se lieraient directement au métal.
Ce n'est qu'en 1893 que la version la plus largement acceptée de la théorie aujourd'hui a été publiée par Alfred Werner . Le travail de Werner comprenait deux changements importants par rapport à la théorie de Blomstrand. Le premier était que Werner a décrit les deux possibilités en termes d'emplacement dans la sphère de coordination. Il a affirmé que si les ions devaient former une chaîne, cela se produirait en dehors de la sphère de coordination tandis que les ions qui se lient directement au métal le feraient à l'intérieur de la sphère de coordination[10]. Dans l'une de ses découvertes les plus importantes, Werner a cependant réfuté la majorité de la théorie de la chaîne. Werner a découvert les arrangements spatiaux des ligands impliqués dans la formation du complexe hexacoordonné de Cobalt. Sa théorie permet de comprendre la différence entre un ligand coordonné et un ion équilibrant la charge dans un composé, par exemple l'ion chlorure dans les chlorures de cobaltammine, et d'expliquer de nombreux isomères auparavant inexplicables.
En 1911, Werner a résolu pour la première fois le complexe de coordination hexol en isomères optiques, renversant la théorie selon laquelle seuls les composés carbonés pouvaient posséder la chiralité[11].
Structures
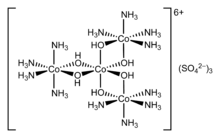
Les ions ou molécules entourant l'atome central sont appelés ligands . Les ligands sont classés comme L ou X (ou une combinaison de ceux-ci), selon le nombre d'électrons qu'ils fournissent pour la liaison entre le ligand et l'atome central. Les ligands L fournissent deux électrons à partir d'une seule paire d'électrons, ce qui donne lieu à une liaison covalente coordonnée . Les ligands X fournissent un électron, l'atome central fournissant l'autre électron, pour ainsi former une liaison covalente régulière. On dit que les ligands sont coordonnés à l'atome. Pour les alcènes, les liaisons pi peuvent se coordonner avec des atomes métalliques. Un exemple est l'éthylène dans le complexe [PtCl3(C2H4)]−.
Géométrie
En chimie de coordination, une structure est d'abord décrite par son numéro de coordination, c'est-à-dire le nombre de ligands attachés au métal (plus précisément, le nombre d'atomes donneurs). Habituellement, on peut compter les ligands attachés, mais parfois même le comptage peut devenir ambigu. Les nombres de coordination sont normalement compris entre deux et neuf, mais il n'est pas rare de compter un grand nombre de ligands pour les lanthanides et les actinides. Le nombre de liaisons dépend de la taille, de la charge et de la configuration électronique de l'ion métallique et des ligands. Les ions métalliques peuvent avoir plus d'un numéro de coordination.
Typiquement, la chimie des complexes de métaux de transition est dominée par les interactions entre les orbitales moléculaires s et p des atomes donneurs dans les ligands et les orbitales d des ions métalliques. Les orbitales s, p et d du métal peuvent accueillir 18 électrons (voir la règle des 18 électrons). Le nombre de coordination maximal pour un certain métal est donc lié à la configuration électronique de l'ion métallique (pour être plus précis, le nombre d'orbitales vides) et au rapport de la taille des ligands et de l'ion métallique. Les grands métaux et les petits ligands conduisent à des nombres de coordination élevés, par exemple [Mo(CN)8]4−. Les petits métaux avec de grands ligands conduisent à de faibles nombres de coordination, par exemple Pt[P(CMe3)]2. En raison de leur grande taille, les lanthanides, les actinides et les premiers métaux de transition ont tendance à avoir des nombres de coordination élevés.
La plupart des structures suivent le modèle de points sur une sphère (par analogie au cas où l'atome central serait au centre d'un polyèdre dont les sommets seraient les emplacements des ligands), dans lequel le chevauchement orbital (entre le ligand et les orbitales métalliques) et les répulsions ligand-ligand tendent à produire certaines géométries régulières. Les géométries les plus observées sont résumées ci-dessous, mais il existe de nombreux cas qui s'écartent d'une géométrie idéale, par exemple en raison de la présence de ligands de différents types (ce qui fait varier la longueur des liaisons et conduit la géométrie à s'écarter du modèle de point sur une sphère), en raison de l'encombrement stérique des ligands ou en raison d'effets électroniques (tels que l'effet Jahn-Teller). Les géométries moléculaires les plus courantes en fonction de la coordinence sont ainsi :
- coordinence = 2 ⇒ géométrie linéaire ;
- coordinence = 3 ⇒ géométrie plane trigonale ;
- coordinence = 4 ⇒ géométrie tétraédrique ou plane carré ;
- coordinence = 5 ⇒ géométrie bipyramidale trigonale ;
- coordinence = 6 ⇒ géométrie octaédrique ;
- coordinence = 7 ⇒ géométrie bipyramidale pentagonale ;
- coordinence = 8 ⇒ géométrie antiprismatique carrée ;
- coordinence = 9 ⇒ géométrie prismatique trigonale tricappée (en).
Les descriptions idéalisées des coordinences 5, 7, 8 et 9 sont souvent indistinctes géométriquement des structures alternatives avec des angles LML (ligand-métal-ligand) légèrement différents, comme entre les structures pyramidales carrées et les structures bipyramidales trigonales[12] :
- coordinence = 5 ⇒ géométrie pyramidale à base carré[13] ;
- coordinence = 7 ⇒ géométrie octaédrique cappée (en) ou prismatique trigonale cappée (en)[14] ;
- coordinence = 8 ⇒ géométrie dodécaédrique (en) ou géométrie prismatique trigonale bicappée (en)[15] ;
- coordinence = 9 ⇒ géométrie antiprismatique carrée cappée (en).
Pour distinguer les coordinations alternatives pour les complexes à cinq coordonnées, l'indice de géométrie τ a été inventé par Addison et al.[16] Cet indice dépend des angles par le centre de coordination et varie entre 0 pour les structures pyramidales carrées et 1 pour les structures bipyramidales trigonales, permettant de classer les cas intermédiaires. Ce système a ensuite été étendu aux complexes à quatre coordonnées par Houser et al.[17] ainsi qu'Okuniewski et al.[18]
Dans les systèmes à faible nombre d'électrons d, en raison d'effets électroniques spéciaux tels que la stabilisation Jahn-Teller (de second ordre)[19], certaines géométries (dans lesquelles les atomes de coordination ne suivent pas un modèle de points sur une sphère) sont stabilisée par rapport aux autres possibilités, par exemple pour certains composés, la géométrie prismatique trigonale est stabilisée par rapport aux structures octaédriques pour la coordinence 6 :
- coordinence = 2 ⇒ géométrie coudée ;
- coordinence = 3 ⇒ géométrie pyramidale trigonale ;
- coordinence = 6 ⇒ géométrie prismatique trigonale.
Isomérie
L'arrangement des ligands est fixe pour un complexe donné, mais dans certains cas, il est susceptible d'être modifié par une réaction qui forme un autre isomère stable.
Il existe de nombreux types d'isomérie dans les complexes de coordination, tout comme dans de nombreux autres composés.
Stéréoisomérie
La stéréoisomérie se produit avec les mêmes liaisons dans des orientations distinctes. La stéréoisomérie peut être classée en[20] :
Isomérie cis-trans et isomérie faciale-méridienne
L'isomérie cis-trans se produit dans les complexes octaédriques et plan carrés (mais pas tétraédriques). Lorsque deux ligands sont adjacents, ils sont dits cis, lorsqu'ils sont opposés, trans . Lorsque trois ligands identiques occupent une face d'un octaèdre, l'isomère est dit facial, ou fac . Dans un isomère fac, deux ligands identiques sont adjacents ou cis l'un à l'autre. Si ces trois ligands et l'ion métallique sont dans un même plan, l'isomère est dit méridien, ou mer . Un isomère mer peut être considéré comme une combinaison d'un trans et d'un cis, car il contient à la fois des paires trans et cis de ligands identiques.
.png.webp) cis-[CoCl2(NH3)4]+
cis-[CoCl2(NH3)4]+.png.webp) trans-[CoCl2(NH3)4]+
trans-[CoCl2(NH3)4]+.png.webp) fac-[CoCl3(NH3)3]
fac-[CoCl3(NH3)3].png.webp) mer-[CoCl3(NH3)3]
mer-[CoCl3(NH3)3]
Isomérie optique
L'isomérie optique se produit lorsqu'un complexe n'est pas superposable avec son image dans un miroir. On l'appelle ainsi parce que les deux isomères sont chacun optiquement actifs, c'est-à-dire qu'ils font tourner le plan de la lumière polarisée dans des directions opposées. Dans la première molécule représentée, le symbole Λ (lambda) est utilisé comme préfixe pour décrire la torsion de l'hélice gauche formée par trois ligands bidentés. La deuxième molécule est l'image miroir de la première, avec le symbole Δ (delta) comme préfixe pour la torsion de l'hélice à droite. Les troisième et quatrième molécules sont une paire similaire d'isomères Λ et Δ, dans ce cas avec deux ligands bidentés et deux ligands monodentés identiques[21].
ferrate(III)-3D-balls.png.webp)
ferrate(III)-3D-balls.png.webp) Δ-[Fe(ox)3]3−
Δ-[Fe(ox)3]3−cobalt(III).png.webp)
cobalt(III).png.webp) Δ-cis-[CoCl2(en)2]+
Δ-cis-[CoCl2(en)2]+
Isomérie structurale
L'isomérie structurelle se produit lorsque les liaisons sont elles-mêmes différentes. On reconnait quatre types d'isomérie structurale : l'isomérie d'ionisation, l'isomérie de solvatation ou d'hydratation, l'isomérie de liaison et l'isomérie de coordination.
- Isomérie d'ionisation – les isomères donnent des ions différents en solution bien qu'ils aient la même composition. Ce type d'isomérie se produit lorsque le contre-ion du complexe est également un ligand potentiel. Par exemple, le sulfate de pentaamminebromocobalt(III) [Co(NH3)5Br]SO4 est violet rouge et en solution donne un précipité avec le chlorure de baryum, confirmant la présence d'ion sulfate, tandis que le bromure de pentaamminesulfatecobalt(III) [Co(NH3)5SO4]Br est rouge et teste négatif pour l'ion sulfate en solution, mais donne à la place un précipité d'AgBr avec du nitrate d'argent[22].
- Isomérie solvatation ou hydratation – les isomères ont la même composition mais diffèrent par le nombre de molécules de solvant servant de ligand vs occupant simplement des sites dans le cristal. Exemples : [Cr(H2O)6]Cl3 est de couleur violette, [CrCl(H2O)5]Cl2·H2O est bleu-vert et [CrCl2(H2O)4]Cl·2H2O est vert foncé. Voir eau de cristallisation[22].
- L'isomérie de liaison se produit avec des ligands de plusieurs types[pas clair] de l'atome donneur, connu sous le nom de ligands ambidentés[23]. Par exemple, le nitrite peut se coordonner via O ou N[24]. Une paire d'isomères de liaison nitrite a des structures (NH3)5CoNO22+ (isomère nitro) et (NH3)5CoONO2+ (isomère nitrite)[23].
- Isomérie de coordination - cela se produit lorsque les ions positifs et négatifs d'un sel sont des ions complexes et que les deux isomères diffèrent dans la distribution des ligands entre le cation et l'anion. Par exemple, [Co(NH3)6][Cr(CN)6] et [Cr(NH3)6][Co(CN)6][22].
Propriétés électroniques
De nombreuses propriétés des complexes de métaux de transition sont dictées par leurs structures électroniques. La structure électronique peut être décrite par un modèle relativement ionique qui attribue des charges formelles aux métaux et aux ligands. Cette approche est l'essence même de la théorie des champs cristallins (CFT). La théorie des champs cristallins, introduite par Hans Bethe en 1929, donne une tentative basée sur la mécanique quantique pour comprendre les complexes. Mais la théorie du champ cristallin traite toutes les interactions dans un complexe comme ioniques et suppose que les ligands peuvent être approximés par des charges ponctuelles négatives.
Des modèles plus sophistiqués englobent la covalence, et cette approche est décrite par la théorie des champs de ligands (LFT) et la théorie des orbites moléculaires (MO). La théorie des champs de ligand, introduite en 1935 et construite à partir de la théorie des orbitales moléculaires, peut gérer une gamme plus large de complexes et peut expliquer les complexes dans lesquels les interactions sont covalentes . Les applications chimiques de la théorie des groupes peuvent aider à la compréhension de la théorie des champs de cristaux ou de ligands, en permettant des solutions simples et basées sur la symétrie aux équations formelles.
Les chimistes ont tendance à utiliser le modèle le plus simple requis pour prédire les propriétés d'intérêt; c'est pourquoi la CFT a été privilégiée pour les discussions lorsque cela était possible. Les théories MO et LF sont plus compliquées, mais offrent une perspective plus réaliste.
La configuration électronique des complexes leur confère des propriétés importantes :
Couleur des complexes de métaux de transition
Les complexes de métaux de transition ont souvent des couleurs spectaculaires causées par des transitions électroniques par l'absorption de la lumière. Pour cette raison, ils sont souvent appliqués comme pigments . La plupart des transitions liées aux complexes métalliques colorés sont soit des transitions d–d, soit des bandes de transfert de charge . Dans la transition ad–d, un électron dans ad orbitale sur le métal est excitée par un photon vers une autre orbitale d d'énergie plus élevée, donc les transitions d–d ne se produisent que pour les complexes d-orbitaux partiellement remplis (d1–9). Pour les complexes ayant une configuration d0 ou d10, le transfert de charge est toujours possible même si les transitions d-d ne le sont pas. Une bande de transfert de charge implique la promotion d'un électron d'une orbitale à base de métal vers une orbitale à base de ligand vide (transfert de charge métal à ligand ou MLCT). L'inverse se produit également : excitation d'un électron dans une orbitale à base de ligand dans une orbitale vide à base de métal (transfert de charge ligand-to-metal ou LMCT). Ces phénomènes peuvent être observés à l'aide de la spectroscopie électronique ; également connu sous le nom d' UV-Vis[25]. Pour les composés simples de symétrie élevée, les transitions d – d peuvent être attribuées à l'aide de diagrammes Tanabe-Sugano. Ces missions sont de plus en plus soutenues par la chimie computationnelle.
| Fe2+ | Fe3+ | Co2+ | Cu2+ | Al3+ | Cr3+ | |
|---|---|---|---|---|---|---|
| Ion hydraté | [Fe(H2O)6]2+ Vert pâle Suspension |
[Fe(H2O)6]3+ Jaune marron Suspension |
[Co(H2O)6]2+ Rose Suspension |
[Cu(H2O)6]2+ Bleu Suspension |
[Al(H2O)6]3+ Incolore Suspension |
[Cr(H2O)6]3+ Vert Suspension |
| (OH)− dilué | [Fe(H2O)4(OH)2] Vert foncé Précipité |
[Fe(H2O)3(OH)3] Marron Précipité |
[Co(H2O)4(OH)2] Bleu vert Précipité |
[Cu(H2O)4(OH)2] Bleu Précipité |
[Al(H2O)3(OH)3] Blanche Précipité |
[Cr(H2O)3(OH)3] Vert Précipité |
| (OH)− concentré | [Fe(H2O)4(OH)2] Vert foncé Précipité |
[Fe(H2O)3(OH)3] Marron Précipité |
[Co(H2O)4(OH)2] Bleu vert Précipité |
[Cu(H2O)4(OH)2] Bleu Précipité |
[Al(OH)4]− Incolore Solution |
[Cr(OH)6]3− Vert Suspension |
| NH3 dilué | [Fe(NH3)6]2+ Vert foncé Précipité |
[Fe(NH3)6]3+ Marron Précipité |
[Co(NH3)6]2+ Couleur paille Suspension |
[Cu(NH3)4(H2O)2]2+ Bleu profond Suspension |
[Al(NH3)3]3+ blanche Précipité |
[Cr(NH3)6]3+ Mauve Suspension |
| NH3 concentré | [Fe(NH3)6]2+ Vert foncé Précipité |
[Fe(NH3)6]3+ Marron Précipité |
[Co(NH3)6]2+ Couleur paille Suspension |
[Cu(NH3)4(H2O)2]2+ Bleu profond Suspension |
[Al(NH3)3]3+ blanche Précipité |
[Cr(NH3)6]3+ Mauve Suspension |
| (CO3)2− | FeCO3 Vert foncé Précipité |
Fe2(CO3)3 Marron Précipité + bulles |
CoCO3 Rose Précipité |
CuCO3 Bleu vert Précipité |
Couleurs des complexes de lanthanides
Superficiellement, les complexes de lanthanides sont similaires à ceux des métaux de transition en ce que certains sont colorés. Cependant, pour les ions communs Ln3+ (Ln = lanthanide) les couleurs sont toutes pâles, et peu influencées par la nature du ligand. Les couleurs sont dues aux transitions d'électrons 4f. Comme les orbitales 4f dans les lanthanides sont "enfouies" dans le noyau de xénon et protégées du ligand par les orbitales 5s et 5p, elles ne sont donc pas influencées par les ligands dans une grande mesure, ce qui conduit à une division du champ cristallin beaucoup plus petite que dans les métaux de transition. . Le spectre d'absorption d'un ion Ln3+ se rapproche de celui de l'ion libre où les états électroniques sont décrits par couplage spin-orbite . Cela contraste avec les métaux de transition où l'état fondamental est divisé par le champ cristallin. Les absorptions pour Ln3+ sont faibles car les transitions dipolaires électriques sont interdites de parité (Laporte interdite) mais peuvent gagner en intensité sous l'effet d'un champ de ligand à faible symétrie ou se mélangeant avec des états électroniques plus élevés (eg orbitales d). ff les bandes d'absorption sont extrêmement nettes ce qui contraste avec celles observées pour les métaux de transition qui ont généralement des bandes larges[26]. Cela peut entraîner des effets extrêmement inhabituels, tels que des changements de couleur importants sous différentes formes d'éclairage.
Magnétisme
Les complexes métalliques qui ont des électrons non appariés sont magnétiques . En ne considérant que les complexes monométalliques, des électrons non appariés apparaissent parce que le complexe a un nombre impair d'électrons ou parce que l'appariement d'électrons est déstabilisé. Ainsi, les espèces monomériques de Ti(III) possèdent un "électron d" et doivent être (para)magnétiques, quelle que soit la géométrie ou la nature des ligands. Ti(II), avec deux électrons d, forme des complexes qui ont deux électrons non appariés et d'autres qui n'en ont aucun. Cet effet est illustré par les composés TiX2[(CH3)2PCH2CH2P(CH3)2]2 : lorsque X = Cl, le complexe est paramagnétique (configuration à haut spin), alors que lorsque X = CH3, il est diamagnétique (configuration bas spin). Il est important de réaliser que les ligands fournissent un moyen important d'ajuster les propriétés de l' état fondamental .
Dans les complexes bimétalliques et polymétalliques, dans lesquels les centres individuels ont un nombre impair d'électrons ou qui sont à spin élevé, la situation est plus compliquée. S'il y a interaction (directe ou via un ligand) entre les deux (ou plusieurs) centres métalliques, les électrons peuvent se coupler (couplage antiferromagnétique, résultant en un composé diamagnétique), ou ils peuvent s'améliorer (couplage ferromagnétique). Lorsqu'il n'y a pas d'interaction, les deux centres métalliques individuels (ou plus) se comportent comme s'ils étaient dans deux molécules distinctes.
Réactivité
Les complexes montrent une variété de réactivités possibles[27] :
- Transferts d'électrons
- Le transfert d'électrons (ET) entre les ions métalliques peut se produire via deux mécanismes distincts, les transferts d'électrons de sphère interne et externe. Dans une réaction de sphère interne, un ligand pontant sert de conduit pour ET.
- Échange de ligands (dégénéré)
- Un indicateur important de la réactivité est le taux d'échange dégénéré de ligands. Par exemple, le taux d'échange d'eau coordonnée dans les complexes [M(H2O)6]n+ varie sur 20 ordres de grandeur. Les complexes où les ligands sont libérés et rebondissent rapidement sont classés comme labiles. De tels complexes labiles peuvent être assez stables thermodynamiquement. Les complexes de métaux labiles typiques ont soit des électrons à faible charge (Na+), dans des orbitales d qui sont antiliants par rapport aux ligands (Zn2+), soit manquent de covalence (Ln3+, où Ln est n'importe quel lanthanide). La labilité d'un complexe métallique dépend également des configurations à haut spin par rapport aux configurations à faible spin lorsque cela est possible. Ainsi, le Fe(II) et le Co(III) à haut spin forment des complexes labiles, tandis que les analogues à bas spin sont inertes. Le Cr(III) ne peut exister que dans l'état de spin bas (quatuor), qui est inerte en raison de son état d'oxydation formel élevé, de l'absence d'électrons dans les orbitales qui sont antiliantes M – L, ainsi que d'une certaine "stabilisation du champ de ligand" associée au configuration d3 .
- Processus associatifs
- Les complexes qui ont des orbitales non remplies ou à moitié remplies montrent souvent la capacité de réagir avec des substrats. La plupart des substrats ont un état fondamental singulet ; c'est-à-dire qu'ils ont des paires d'électrons isolés (par exemple, de l'eau, des amines, des éthers), de sorte que ces substrats ont besoin d'une orbitale vide pour pouvoir réagir avec un centre métallique. Certains substrats (par exemple, l'oxygène moléculaire) ont un état fondamental triplet, ce qui fait que les métaux avec des orbitales à moitié remplies ont tendance à réagir avec de tels substrats (il faut dire que la molécule de dioxygène a aussi des paires isolées, donc elle est également capable réagir comme une base de Lewis "normale").
Si les ligands autour du métal sont soigneusement choisis, le métal peut aider aux transformations (stœchiométriques ou catalytiques) des molécules ou être utilisé comme capteur.
Classification
Les complexes métalliques, également connus sous le nom de composés de coordination, comprennent pratiquement tous les composés métalliques[28]. L'étude de la "chimie de coordination" est l'étude de la "chimie inorganique" de tous les métaux alcalins et alcalino-terreux, des métaux de transition, des lanthanides, des actinides et des métalloïdes. Ainsi, la chimie de coordination est la chimie de la majorité du tableau périodique. Les métaux et les ions métalliques existent, du moins dans les phases condensées, uniquement entourés de ligands.
Les domaines de la chimie de coordination peuvent être classés selon la nature des ligands, en termes généraux :
- Classique (ou "complexes de Werner ") : les ligands de la chimie de coordination classique se lient aux métaux, presque exclusivement, via leurs doublets non liants résidant sur les atomes du groupe principal du ligand. Les ligands typiques sont H2O, NH3, Cl−, CN−, en. Certains des membres les plus simples de ces complexes sont décrits comme complexes aqua et ammine.
- Exemples : [Co(EDTA)]−, [Co(NH3)6]3+, [Fe(C2O4)3]3−.
- Chimie organométallique : les ligands sont organiques (alcènes, alcynes, alkyles) ainsi que des ligands « de type organique » tels que les phosphines, les hydrures et le CO.
- Exemple : [(η5-C5H5)Fe(CO)(µ-CO)]2.
- Chimie bioinorganique : Les ligands sont d'origine naturelle, comprenant notamment les chaînes latérales des acides aminés, et de nombreux cofacteurs tels que les porphyrines, par exemple :
- l'hémoglobine contient de l'hème, complexe de porphyrine et de fer ;
- la chlorophylle contient un complexe de porphyrine et de magnésium ;
- de nombreux ligands naturels sont "classiques", notamment l'eau.
- Chimie des groupes : les ligands comprennent tout ce qui précède ainsi que d'autres ions ou atomes métalliques :
- Exemple : Ru3(CO)12.
- Dans certains cas, il existe des combinaisons de différents champs :
- Exemple : [Fe4S4(Cystéinyl-S)4]2−, dans lequel un groupe est inclus dans une espèce biologiquement active.
La minéralogie, la science des matériaux et la chimie de l'état solide – tels qu'elles s'appliquent aux ions métalliques – sont des sous-ensembles de la chimie de coordination dans le sens où les métaux sont entourés de ligands. Dans de nombreux cas, ces ligands sont des oxydes ou des sulfures, mais les métaux sont néanmoins coordonnés, et les principes et directives discutés ci-dessous s'appliquent. Dans les hydrates, certains ligands sont des molécules d'eau de cristallisation. Il est vrai que l'objectif de la minéralogie, de la science des matériaux et de la chimie de l'état solide diffère de l'orientation habituelle de la coordination ou de la chimie inorganique. Les premières s'intéressent principalement les structures polymères, propriétés résultant des effets collectifs de nombreux métaux fortement interconnectés. En revanche, la chimie de coordination se concentre sur la réactivité et les propriétés des complexes contenant des atomes métalliques individuels ou de petits ensembles d'atomes métalliques.
Nomenclature des complexes de coordination
La méthode de base pour nommer un complexe est la suivante :
- lors de la désignation d'un ion complexe, les ligands sont nommés avant l'ion métallique ;
- le nom des ligands est donné par ordre alphabétique. Les préfixes numériques n'affectent pas l'ordre :
- les ligands monodentés à occurrence multiple reçoivent un préfixe en fonction du nombre d'occurrences : di-, tri-, tétra-, penta- ou hexa-;
- les ligands polydentés multiples (par exemple, éthylènediamine, oxalate) reçoivent bis-, tris-, tétrakis-, etc. ;
- les anions se terminent par o. Cela remplace le « e » final lorsque l'anion se termine par « -ure », « -ate » ou « -ite », par exemple le chlorure devient chloruro et le sulfate devient sulfato. Auparavant, '-ure' était remplacé par '-o' (par exemple chloro et cyano), mais cette règle a été modifiée dans les recommandations IUPAC de 2005 et les formes correctes pour ces ligands sont à présent chloruro et cyanuro[29] ;
- les ligands neutres reçoivent leur nom usuel, à quelques exceptions près : NH3 devient ammine (avec deux « m ») ; H2O devient aqua ; CO devient carbonyle ; NO devient nitrosyle ;
- on écrit ensuite le nom de l'atome ou de l'ion central. Si le complexe est un anion, le nom de l'atome central se terminera par -ate, et son nom latin sera utilisé s'il est disponible (sauf pour le mercure) ;
- l'état d'oxydation de l'atome central doit être spécifié (lorsqu'il est l'un de plusieurs possibles, ou zéro), et doit être écrit sous la forme d'un chiffre romain (ou 0) entre parenthèses ;
- le nom du cation doit être précédé du nom de l'anion.
Exemples:
| métal | changé en |
|---|---|
| cobalt | cobaltate |
| aluminium | aluminate |
| chrome | chromate |
| vanadium | vanadate |
| cuivre | cuprate |
| fer | ferrate |
- [Cd(CN)2(en)2] → dicyanurobis(éthylènediamine)cadmium(II) ;
- [CoCl(NH3)5]SO4 → sulfate de pentaamminechlorurocobalt(III) ;
- [Cu(H2O)6]2+ → ion hexaaquacuivre(II) ;
- [CuCl5NH3]3− → ion amminepentachlorurocuprate(II) ;
- K4[Fe(CN)6] → hexacyanuroferrate de potassium(II) ;
- [NiCl4]2− → ion tétrachloruronickélate(II) (l'utilisation de chloro- a été supprimée de la convention de dénomination IUPAC)[29].
La coordinence des ligands liés à plus d'un centre métallique (ligands pontants) est indiqué par un indice au symbole grec μ placé devant le nom du ligand. Ainsi le dimère de trichlorure d'aluminium est décrit par Al2Cl4(μ2-Cl)2 (en pratique, l'indice est omis lorsqu'il vaut 2).
Tout groupe anionique peut être stabilisé électroniquement par n'importe quel cation. Un complexe anionique peut être stabilisé par un cation hydrogène, devenant un complexe acide qui peut se dissocier pour libérer l'hydrogène cationique. Ce type de composé complexe a un nom avec "ic" ajouté après le métal central. Par exemple, H2[Pt(CN)4] porte le nom d'acide tétracyanoplatinique(II).
Constante de stabilité
L'affinité des ions métalliques pour les ligands est décrite par une constante de stabilité, également appelée constante de formation, et est représentée par le symbole Kf . C'est la constante d'équilibre pour laquelle le métal constitutif se lie à des ligands, et peut être calculée en conséquence, comme dans l'exemple suivant pour un cas simple :
- x M(aq) + y L(aq) z Z(aq)


où : x, y et z sont les coefficients stoechiométriques de chaque espèce. M signifie métal/ion métallique, le L pour les bases de Lewis et enfin Z pour les ions complexes. Les constantes de formation varient considérablement. Des valeurs élevées indiquent que le métal a une forte affinité pour le ligand, à condition que le système soit à l'équilibre[30].
Parfois, la constante de stabilité se présente sous une forme différente, appelée constante de dissociation. Cette constante est exprimée comme l'inverse de la constante de formation et est notée Kd = 1/Kf[31]. Cette constante représente la réaction inverse, c'est la dissociation d'un ion complexe en ses composants métalliques et ligands individuels. Lorsque l'on compare les valeurs de K d, plus la valeur est grande, plus l'ion complexe est instable.
En raison de la formation de ces ions complexes dans les solutions, ils peuvent également jouer un rôle clé dans la solubilité d'autres composés. Lorsqu'un ion complexe se forme, il peut modifier les concentrations de ses composants dans la solution. Par exemple:
- Ag(aq)+ + 2 NH4OH(aq) Ag(NH3)2+ + H2O
- AgCl(s) + H2O(l) Ag(aq)+ + Cl(aq)−.
Si ces réactions se produisaient toutes deux dans le même récipient de réaction, la solubilité du chlorure d'argent serait augmentée par la présence de NH4OH parce que la formation du complexe Diammine argentum(I) consomme une partie importante des ions argent libres de la solution. Selon le principe de Le Chatelier, cela provoque le déplacement vers la droite de la réaction d'équilibre pour la dissolution du chlorure d'argent, qui a pour produit l'ion argent.
Cette nouvelle solubilité peut être calculée à partir des valeurs de Kf et Ksp pour les réactions originales. La solubilité est trouvée essentiellement en combinant les deux équilibres séparés en une seule réaction d'équilibre combinée et cette réaction combinée est celle qui détermine la nouvelle solubilité. Ainsi Kc, la nouvelle constante de solubilité, est désignée par :

Application des composés de coordination
Les métaux n'existent en solution que sous la forme de complexes de coordination. Cette classe de composés est par conséquent, utile dans une grande variété de domaines.
Chimie bioinorganique
En chimie bioinorganique et en chimie bioorganométallique, les complexes de coordination remplissent des fonctions structurelles ou catalytiques. On estime que 30 % des protéines contiennent des ions métalliques. Les exemples, citons la vitamine B12 de couleur intense, le groupe hème de l'hémoglobine, les cytochromes, le groupe chlore de la chlorophylle et la carboxypeptidase, une enzyme hydrolytique importante dans la digestion. Une autre enzyme à ions complexes est la catalase, qui décompose le peroxyde d'hydrogène, déchet de la cellule . Les composés de coordination synthétiques sont également utilisés pour se lier aux protéines et en particulier aux acides nucléiques (par exemple, le cisplatine, un médicament anticancéreux).
Industrie
La catalyse homogène est une application majeure des composés de coordination pour la production de substances organiques. Les procédés comprennent l'hydrogénation, l' hydroformylation, l'oxydation . Dans un exemple, une combinaison de trichlorure de titane et de triéthylaluminium donne naissance à des catalyseurs Ziegler-Natta, utilisés pour la polymérisation de l'éthylène et du propylène pour donner des polymères d'une grande importance commerciale les fibres, les films et les plastiques.
Le nickel, le cobalt et le cuivre peuvent être extraits à l'aide de procédés hydrométallurgiques faisant intervenir des ions complexes. Ils sont extraits de leurs minerais sous forme de complexes d'amines . Les métaux peuvent également être séparés en utilisant la précipitation sélective et la solubilité des ions complexes. Le cyanure est principalement utilisé pour extraire l'or et l'argent de leurs minerais.
Les complexes de phtalocyanine sont une classe importante de pigments.
Analyse
À une certaine époque, des composés de coordination ont été utilisés pour identifier la présence de métaux dans un échantillon. L'analyse inorganique qualitative a été largement remplacée par des méthodes d'analyse instrumentales telles que la spectroscopie d'absorption atomique (AAS), la spectroscopie d'émission atomique à plasma à couplage inductif (ICP-AES) et la spectrométrie de masse à plasma à couplage inductif (ICP-MS).
Notes et références
- Geoffrey A. Lawrance, Introduction to Coordination Chemistry, Wiley, (ISBN 9780470687123, DOI 10.1002/9780470687123)
- (en) « complex », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8)
- (en) « coordination entity », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8)
- (en) N. N. Greenwood et A. Earnshaw, Chemistry of the Elements, 2e éd., Butterworth-Heinemann, 1997. (ISBN 978-0-08-037941-8)
- « Definition of coordination sphere », chemistry-dictionary.com
- « What Is A Coordination Compound? », Purdue University Department of Chemistry
- Frank Albert Cotton, Geoffrey Wilkinson et Carlos A. Murillo, Advanced Inorganic Chemistry, (ISBN 978-0-471-19957-1), p. 1355
- Gary L. Miessler et Donald Arthur Tarr, Inorganic Chemistry, (ISBN 978-0-13-841891-5), p. 642
- (en) « Coordination compound - History of coordination compounds », Encyclopedia Britannica (consulté le )
- « Coordination Compound »
- (de) Werner, « Zur Kenntnis des asymmetrischen Kobaltatoms. I », Berichte der Deutschen Chemischen Gesellschaft, vol. 44, no 2, , p. 1887–1898 (DOI 10.1002/cber.19110440297, lire en ligne)
- Wells A.F. (1984) Structural Inorganic Chemistry 5th edition Oxford Science Publications (ISBN 0-19-855370-6)
- Angelo R. Rossi et Roald. Hoffmann, « Transition metal pentacoordination », Inorganic Chemistry, vol. 14, no 2, , p. 365–374 (DOI 10.1021/ic50144a032)
- Roald. Hoffmann, Barbara F. Beier, Earl L. Muetterties et Angelo R. Rossi, « Seven-coordination. A molecular orbital exploration of structure, stereochemistry, and reaction dynamics », Inorganic Chemistry, vol. 16, no 3, , p. 511–522 (DOI 10.1021/ic50169a002)
- Jeremy K. Burdett, Roald Hoffmann et Robert C. Fay, « Eight-Coordination », Inorganic Chemistry, vol. 17, no 9, , p. 2553–2568 (DOI 10.1021/ic50187a041)
- Addison, Rao, Reedijk et van Rijn, « Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate », J. Chem. Soc., Dalton Trans., no 7, , p. 1349–1356 (DOI 10.1039/dt9840001349)
- Yang, Powell et Houser, « Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4 », Dalton Trans., no 9, , p. 955–64 (PMID 17308676, DOI 10.1039/b617136b)
- Okuniewski, Rosiak, Chojnacki et Becker, « Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas », Polyhedron, vol. 90, , p. 47–57 (DOI 10.1016/j.poly.2015.01.035)
- Kaupp, « "Non-VSEPR" Structures and Bonding in d0 Systems », Angew. Chem. Int. Ed. Engl., vol. 40, no 1, , p. 3534–3565 (DOI 10.1002/1521-3773(20011001)40:19<3534::AID-ANIE3534>3.0.CO;2-#)
- von Zelewsky, A. "Stereochemistry of Coordination Compounds" John Wiley: Chichester, 1995. (ISBN 0-471-95599-X).
- Gary L. Miessler et Donald Arthur Tarr, Inorganic Chemistry, , 315, 316 (ISBN 978-0-13-841891-5), « 9 »
- Huheey, James E., Inorganic Chemistry (3rd ed., Harper & Row 1983), p. 524–5 (ISBN 0-06-042987-9)
- William L. Jolly, Modern Inorganic Chemistry, McGraw-Hill, , 357–9 p. (ISBN 0-07-032760-2)
- Huheey, James E., Inorganic Chemistry (3rd ed., Harper & Row 1983), p. 513–24 (ISBN 0-06-042987-9)
- D. Harris et M. Bertolucci, Symmetry and Spectroscopy, Dover Publications, (ISBN 9780486661445)
- Simon Cotton, Lanthanide and Actinide Chemistry, John Wiley & Sons Ltd,
- R. G. Wilkins Kinetics and Mechanism of Reactions of Transition Metal Complexes, 2nd Edition, VCH, Weinheim, 1991. (ISBN 1-56081-125-0)
- Exception: metal vapors, plasmas, and alloys.
- « Nomenclature of Inorganic Chemistry IUPAC Recommendations 2005 » [archive du ], IUPAC (consulté le )
- « Complex Ion Equilibria »
- Stretton, « Solubility and Complex-ion Equilibria »
Voir également
Articles connexes
- Complexe activé
- Géométrie de coordination
- Composés d'inclusion
- La chimie organométallique traite d'une classe spéciale de composés de coordination où des fragments organiques sont liés à un métal au moins par un atome de carbone.
Liens externes
- Ressources relatives à la santé :
- (en) Medical Subject Headings
- (cs + sk) WikiSkripta
- Notices dans des dictionnaires ou encyclopédies généralistes :
 Portail de la chimie
Portail de la chimie