Dimère de (cyclopentadiényl)fer dicarbonyle
Le dimère de (cyclopentadiényl)fer dicarbonyle, est un complexe organoferrique (en) de formule chimique [(η5-C5H5)Fe(CO)(µ-CO)]2, souvent abrégée Cp2Fe2(CO)4. Parfois symbolisé Fp2 ou appelé « dimère fip », il s'agit d'un solide cristallisé de couleur sombre rougeâtre, très soluble dans les solvants organiques faiblement polaires comme le chloroforme CHCl3 et la pyridine C5H5N, mais moins soluble dans le tétrachlorométhane CCl4 et le disulfure de carbone CS2. Il est insoluble dans l'eau mais reste stable au contact de l'eau. Il est relativement stable au contact de l'air et peut être conservé comme matière première pour produire des dérivés du (cyclopentadiényl)fer dicarbonyle (η5-C5H5)Fe(CO)2 moins stables[3].
| Dimère de (cyclopentadiényl)fer dicarbonyle | |||
| |||
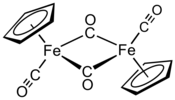
| Structure du dimère trans de (cyclopentadiényl)fer dicarbonyle | |||
| Identification | |||
|---|---|---|---|
| No CAS | |||
| No ECHA | 100.032.057 | ||
| No CE | 235-276-3 | ||
| PubChem | 11110989 | ||
| SMILES | |||
| InChI | |||
| Apparence | solide brun rougeâtre inodore[1] | ||
| Propriétés chimiques | |||
| Formule | C14H10Fe2O4 |
||
| Masse molaire[2] | 353,917 ± 0,017 g/mol C 47,51 %, H 2,85 %, Fe 31,56 %, O 18,08 %, |
||
| Propriétés physiques | |||
| T° fusion | 194 °C[1] | ||
| Précautions | |||
| SGH[1] | |||
 Attention |
|||
| NFPA 704[1] | |||
| Transport[1] | |||
|
|||
| Unités du SI et CNTP, sauf indication contraire. | |||


Synthèse et structure
On prépare le dimère de (cyclopentadiényl)fer dicarbonyle comme lors de sa première caractérisation, en faisant réagir du pentacarbonyle de fer Fe(CO)5 avec du dicyclopentadiène C10H12[4] :
Cela permet le craquage du dicyclopentadiène pour donner le cyclopentadiène C5H6, lequel réagit avec le pentacarbonyle de fer en libérant le monoxyde de carbone. Des voies photochimiques et thermiques ont été affinées par la suite, avec un hydrure intermédiaire[5]. Cette méthode est utilisée dans l'enseignement au laboratoire[6].
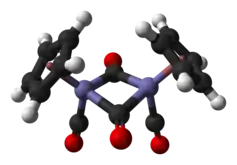
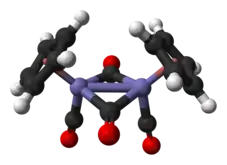
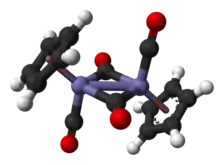
En solution, le Cp2Fe2(CO)4 peut être vu comme un dimère de complexes en tabouret de piano. Il existe sous trois isomères : une forme cis, une forme trans et une forme sans ligand pontant, dite « ouverte ». Les deux premières sont unies par deux ligands CO pontants et ont pour formule [(η5-C5H5)Fe(CO)(µ-CO)]2, tandis que la dernière n'a pas de ligand pontant mais est unie par une liaison Fe–Fe selon la formule (η5-C5H5)(CO)2Fe–Fe(CO)2(η5-C5H5) ce qui permet l'interconversion entre les les formes cis et trans, qui sont prépondérantes à l'équilibre ; les cycles cyclopentadiényle sont situés du même côté du plan défini par les ligands CO pontants dans la forme cis, et de part et d'autre de ce plan dans la forme trans :

On sait par ailleurs que les ligands carbonyle échangent leur position : ceux de l'isomère trans peuvent s'intervertir entre CO pontant et CO terminal aussi bien en passant par l'isomère ouvert que par torsion, tandis que ceux de l'isomère cis ne peuvent s'échanger qu'en passant par l'isomère ouvert[7].
En solution, les isomères cis, trans et ouverts s'interconvertissent rapidement à température ambiante, faisant de la structure une molécule fluxionnelle (en). Ces échanges sont plus rapides que l'échelle de temps de la RMN, de sorte qu'on observe un signal moyen unique du cyclopentadiényle en RMN 1H à 25 °C. De même, le spectre par RMN 13C présente un signal net du carbonyle au-dessus de −10 °C, tandis que le signal du cyclopentadiényle s'affine en un seul pic au-dessus de 60 °C. Les analyses par RMN montrent que l'isomère cis est légèrement plus abondant que l'isomère trans à température ambiante, tandis que la forme ouverte reste peu abondante[7]. Les interconversions sont en revanche moins rapides que l'échelle de temps de la spectroscopie infrarouge, de sorte qu'on observe trois absorptions pour chaque isomère. Les ligands CO pontants apparaissent vers 1 780 cm−1 tandis que les CO terminaux sont observés vers 1 980 cm−1[6]. La structure moyenne de ces isomères correspond à un moment dipolaire de 3,1 D[8].
La structure moléculaire à l'état solide des isomères cis et trans a été analysée par diffraction aux rayons X et par diffraction de neutrons. La distance Fe–Fe et la longueur des liaisons C–Fe sont identiques dans les rhomboïdes Fe2C2, un cycle à quatre sommets parfaitement plan dans l'isomère trans contre un rhomboïde replié avec un angle de 164° dans l'isomère cis et des distorsions importantes dans le cycle cyclopentadiényle de l'isomère trans reflétant différentes populations orbitales dans le ligand cyclopentadiényle[5].

Contrairement aux première représentations qui figuraient une liaison entre les deux atomes de fer, les analyses théoriques indiquent l'absence de liaison Fe–Fe dans les isomères cis et trans. Ce point de vue est conforme aux calculs et aux données obtenues par cristallographie aux rayons X, qui indiquent que la densité électronique est insuffisante pour une liaison entre les atomes de fer[9]. Ce point reste cependant débattu, enssentiellement à partir de la réactivité chimique et des données spectroscopiques, la densité électronique n'étant pas nécessairement la meilleure indication de l'existence d'une liaison chimique, tandis que les implications de la règle des 18 électrons sur le comportement chimique et spectroscopique des groupes carbonyle ne seraient pas cohérentes avec l'absence de liaison entre les atomes de fer[10].
- Représentation des isomères cis et trans avec et sans liaison Fe–Fe

Réactions
Bien qu'il n'ait pas de valeur commerciale particulière, le dimère de (cyclopentadiényl)fer dicarbonyle est en enjeu en chimie organométallique car il est peu coûteux et ses dérivés halogénure et organiques sont robustes.
Le clivage réducteur par un métal alcalin, ou une forme activée de ce dernier, de [CpFe(CO)2]2, formellement un complexe de fer(I), produit des dérivés de métaux alcalins formellement dérivés de l'anion (cyclopentadiényl)fer dicarbonyle [CpFe(CO)2]−, écrit Fp−, formellement fer(0), qui sont supposés exister sous la forme d'une paire d'ions de contact. Le sodium élémentaire et l'amalgame de sodium sont des réducteurs typiques[11] ; on a également utilisé pour ce faire un alliage NaK, du graphite de potassium KC8 et des trialkylborohydrures de métaux alcalins, comme ci-dessous le triéthylborohydrure de potassium K[(C2H5)3BH] :
- [CpFe(CO)2]2 + 2 Na ⟶ 2 Na[CpFe(CO)2] ;
- [CpFe(CO)2]2 + 2 K[(C2H5)3BH] ⟶ 2 K[CpFe(CO)2] + H2 + 2 (C2H5)3B.
Le Na[CpFe(CO)2] est très étudié car il est facilement l'objet d'alkylations, d'acylations ou de métallations par traitement avec des électrophiles appropriés[12]. C'est un excellent nucléophile SN2, un à deux ordres de grandeur plus nucléophile que le thiophénolate C6H5S− lorsqu'il réagit avec des bromures d'alkyle primaires et secondaires[13].
Le traitement de NaFp avec un halogénoalcane (iodure ou bromure) donne un dérivé alkylé du complexe.
Fp2 peut également être clivé avec des métaux alcalins[14] et par réduction électrochimique[15],[16].
L'oxydation par les halogènes clive [CpFe(CO)2]2 pour donner les halogénures FpX correspondants, avec le fer formellement à l'état d'oxydation +2 ; X ci-dessous représente le chlore, le brome ou l'iode :
- [CpFe(CO)2]2 + X2 ⟶ 2 CpFe(CO)2X.
L'un de ces produits est l'iodure de (cyclopentadiényl)fer dicarbonyle (η5-C5H5)Fe(CO)2I.
La réaction des halogénures FpX avec des alcènes, des alcynes et des ligands labiles neutres en présence d'accepteurs d'anions halogénure tels que le bromure d'aluminium Al2Br6 et le tétrafluoroborate d'argent AgBF4 donne les complexes Fp+ correspondants[17]. On obtient des sels du complexe [Fp(isobutylène)]+ par réaction de NaFp avec du chlorure de méthallyle CH2=C(CH3)CH2Cl suivie d'une protonolyse (en). Ces sels sont des précurseurs polyvalents et pratiques vers d'autres complexes cationiques Fp–alcène et Fp–alcyne[18]. L'échange est facilité par l'élimination de l'isobutylène HC(CH3)3, à la fois gazeux et encombrant[19]. D'une manière générale, les alcènes les moins substitués se lient le plus fortement et peuvent déplacer des ligands alcène davantage altérés.
Les complexes d'alcènes et d'alcynes peuvent également être obtenus en chauffant des complexes aqua ou cationiques, comme le complexe [Fp(thf)]+BF4−, avec l'alcène ou l'alcyne[20]. Les complexes [FpL]+BF4− peuvent également être préparés en traitant du FpMe avec HBF4·Et2O dans le dichlorométhane CH2Cl2 à −78 °C, suivi par l'addition du ligand L[21]. Il est également possible de préparer les complexes Fp–alcène de manière indirecte à partir de l'anion. L'élimination de l'hydrure des complexes Fp–alkyle à l'aide d'hexafluorophosphate de triphénylméthyle (en) (C6H5)3CPF6 donne ainsi des complexes [Fp(α-alcène)]+.
La réaction de NaFp avec un époxyde suivie par la déshydratation sous l'effet d'un acide permet également de produire des complexes d'alcènes. Les complexes Fp(alcène)+ sont stables par rapport à la bromation, l'hydrogénation et l'oxymercuration (en), mais l'alcène est facilement libéré par l'iodure de sodium dans l'acétone ou par chauffage avec de l'acétonitrile[22]. Le ligand alcène de ces cations est activé pour les substitutions nucléophiles, ce qui ouvre la voie à de nombreuses réactions établissant des liaisons C–C. Les additions nucléophiles surviennent généralement sur les atomes de carbone les plus substitués. Cette régiosélectivité, le plus souvent limitée, est généralement attribuée à la densité de charges positives plus élevée à ce niveau. L'addition du nucléophile est entièrement stéréosélective, en position anti par rapport au groupe Fp.
exchange.png.webp)
Les complexes Fp(alcyne)+ analogues connaissent également des additions nucléophiles sous l'effet de divers nucléophiles à carbone, azote ou oxygène[23]. Les complexes π Fp(alcène)+ et Fp(alcyne)+ sont également assez acides au niveau respectivement des positions allyliques et propargyliques et peuvent être déprotonés avec des amines basiques telles que la triéthylamine Et3N pour donner les complexes σ neutres Fp–allyl et Fp–allényl[18] :
- Fp(η2-H2C=CHCH2CH3)+BF4− + Et3N ⟶ Fp–CH2CH=CHCH3 + Et3NH+BF4−.
Ces complexes Fp–allyl et Fp–allényl peuvent réagir avec des électrophiles cationiques E, tels que l'oxonium Me3O+, des carbocations, ou des ions oxocarbenium (en), pour réaliser des fonctionnalisations allyliques et propargyliques[18]. On a pu montrer que le complexe apparenté [Cp*Fe(CO)2(thf)]+[BF4]−, dans lequel Cp* désigne le ligand pentaméthylcyclopentadiényle (CH3)5C5 peut catalyser la fonctionnalisation C–H allylique et propargylique en combinant fonctionalisations électrophiles et déprotonations[24].
- Fp–CH2CH=CHCH3 + E+BF4− ⟶ Fp(η2-H2C=CHCH(E)CH3)+BF4−.
Les complexes η2-allényle du cation Fp+ et de cations (cyclopentadiényl)fer dicarbonyle substitués ont également été caractérisés, la cristallographie aux rayons X montrant une incurvation significative au niveau de l'atome de carbone allénique central (angle de liaison inférieur à 150°)[25],[26].
Des réactifs à base de Fp ont été développés pour la cyclopropanation[27]. Le réactif principal est produit à partir de FpNa avec du thioéther et d'iodométhane CH3I, qui se conservent mieux que les intermédiaires de réactions de Simmons-Smith et les diazoalcanes.
L'utilisation de [FpCH2S(CH3)2]BF4 ne demande pas de conditions opératoires très particulières. L'addition de trichlorure de fer FeCl3 permet d'éliminer les sous-produits.
Des précurseurs de Fp=CH2+, comme le FpCH2OMe converti en carbène de fer par protonation, ont également été employés comme réactifs de cyclopropanation[28].
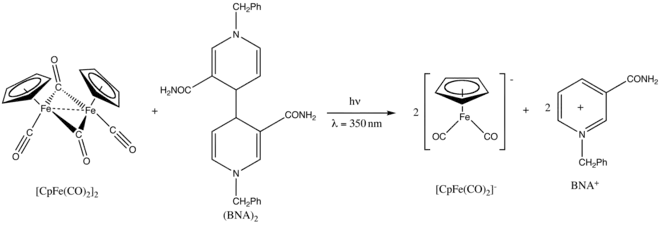
Fp2 connaît également des réactions photochimiques[29]. Il est par exemple réduit par le dimère de 1-benzyl-1,4-dihydronicotinamide, également noté (BNA)2, sous l'effet d'un rayonnement ultraviolet à 350 nm[30].
Notes et références
- « Fiche du composé Bis(dicarbonylcyclopentadienyliron), 99% », sur Alfa Aesar (consulté le ).
- Masse molaire calculée d’après « Atomic weights of the elements 2007 », sur www.chem.qmul.ac.uk.
- (en) William J. Kelly, « Bis(dicarbonylcyclopentadienyliron) », Encyclopedia of Reagents for Organic Synthesis, (DOI 10.1002/047084289X.rb139, lire en ligne)
- (en) T. S. Piper, F. A. Cotton et G.Wilkinson, « Cyclopentadienyl-carbon monoxide and related compounds of some transitional metals », Journal of Inorganic and Nuclear Chemistry, vol. 1, no 3, , p. 165-174 (DOI 10.1016/0022-1902(55)80053-X, lire en ligne)
- (en) G. Wilkinson, Comprehensive Organometallic Chemistry, vol. 4, Pergamon Press, 1982, p. 513–613. (ISBN 978-0-08-025269-8)
- (en) G. Girolami, T. Rauchfuss et R. Angelici, Synthesis and Technique in Inorganic Chemistry, 3e éd., University Science Books, 1999, p. 171–180. (ISBN 978-0-935702-48-4)
- (en) Daniel C. Harris, Edward Rosenberg et John D. Roberts, « Carbon-13 nuclear magnetic resonance spectra and mechanism of bridge–terminal carbonyl exchange in di-µ-carbonyl-bis[carbonyl(η-cyclopentadienyl)iron](Fe–Fe)[{(η-C5H5)Fe(CO)2}2]; cd-di-µ-carbonyl-f-carbonyl-ae-di(η-cyclopentadienyl)-b-(triethyl -phosphite)di-iron(Fe–Fe)[(η-C5H5)2Fe2(CO)3P(OEt)3], and some related complexes », Journal of the Chemical Society, Dalton Transactions, vol. 1974, no 22, , p. 2398-2403 (DOI 10.1039/DT9740002398, lire en ligne)
- (en) F. Albert Cotton et G. Yagupsky, « Tautomeric changes in metal carbonyls. I. π-Cyclopentadienyliron dicarbonyl dimer and π-cyclopentadienyl-ruthenum dicarbonyl dimer », Inorganic Chemistry, vol. 6, no 1, , p. 15-20 (DOI 10.1021/ic50047a005, lire en ligne)
- (en) Jennifer C. Green, Malcolm L. H. Green et Gerard Parkin, « The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds », Chemical Communications, vol. 48, no 94, , p. 11481-11503 (PMID 23047247, DOI 10.1039/c2cc35304k, lire en ligne)
- (en) Jay A. Labinger, « Does cyclopentadienyl iron dicarbonyl dimer have a metal–metal bond? Who’s asking? », Inorganica Chimica Acta, vol. 424, , p. 14-19 (DOI 10.1016/j.ica.2014.04.022, lire en ligne)
- (en) Tony C. T. Chang, Myron Rosenblum et Nancy Simms, « Vinylation of enolates with a vinyl cation equivalent: trans-3-methyl-2-vinylcyclohexanon », Organic Syntheses, vol. 66, , p. 95 (DOI 10.15227/orgsyn.066.0095, lire en ligne)
- (en) Robert Bruce King, « Applications of metal carbonyl anions in the synthesis of ususual organometallic compounds », Accounts of Chemical Research, vol. 3, no 12, , p. 417-427 (DOI 10.1021/ar50036a004, lire en ligne)
- (en) Raymond E. Dessy, Rudolph L. Pohl et R. Bruce King, « Organometallic Electrochemistry. VII. The Nucleophilicities of Metallic and Metalloidal Anions Derived from Metals of Groups IV, V, VI, VII, and VIII », Journal of the American Chemical Society, vol. 88, no 22, , p. 5121-5124 (DOI 10.1021/ja00974a015, lire en ligne)
- (en) John E. Ellis et Elroy A. Flom, « The chemistry of metal carbonyl anions : III. Sodium-potassium alloy: An efficient reagent for the production of metal carbonyl anions », Journal of Organometallic Chemistry, vol. 99, no 2, , p. 263-268 (DOI 10.1016/S0022-328X(00)88455-7, lire en ligne)
- (en) Raymond E. Dessy, R. Bruce King et Morgan Waldrop, « Organometallic Electrochemistry. V. The Transition Series », Journal of the American Chemical Society, vol. 88, no 22, , p. 5112-5117 (DOI 10.1021/ja00974a013, lire en ligne)
- (en) Raymond E. Dessy, Paul M. Weissman et Rudolph L. Pohl, « Organometallic Electrochemistry. VI. Electrochemical Scission of Metal-Metal Bonds », Journal of the American Chemical Society, vol. 88, no 22, , p. 5117-5121 (DOI 10.1021/ja00974a014, lire en ligne)
- (en) J. Silver, Chemistry of Iron, Springer Netherlands, 1993. (ISBN 978-9401121408) (OCLC 840309324)
- (en) A. Cutler, D. Ehnholt, P. Lennon, K. Nicholas, David F. Marten, M. Madhavarao, S. Raghu, A. Rosan et M. Rosenblum, « Chemistry of dicarbonyl η5-cyclopentadienyliron complexes. General syntheses of monosubstituted η2-olefin complexes and of 1-substituted η1-allyl complexes. Conformational effects on the course of deprotonation of (η2-olefin) cations », Journal of the American Chemical Society, vol. 97, no 11, , p. 3149-3157 (DOI 10.1021/ja00844a038, lire en ligne)
- (en) Mark M. Turnbull, « Dicarbonyl(cyclopentadienyl)(isobutene)iron Tetrafluoroborate », Encyclopedia of Reagents for Organic Synthesis, (DOI 10.1002/047084289X.rd080, lire en ligne)
- (en) D. F. Schriver, M. I. Bruce et G. Wilkinson, Iron, Ruthenium and Osmium, Elsevier Science, 1995. (ISBN 978-0-08-096396-9) (OCLC 953660855)
- (en) Mark D. Redlich, Michael F. Mayer et M. Mahmun Hossain, « Iron Lewis acid [η5-C5H5)Fe(CO) 2(THF)]+ catalyzed organic reactions », Aldrichimica Acta, vol. 36, no 1, , p. 3-13
- (en) A. J. Pearson, Iron Compounds in Organic Synthesis, Academic Press, 1994. p. 22–35. (ISBN 978-0-12-548270-7)
- (en) Munetaka Akita, Satoshi Kakuta, Shuichiro Sugimoto, Masako Terada, Masako Tanaka et Yoshihiko Moro-oka, « Nucleophilic Addition to the η2-Alkyne Ligand in [CpFe(CO)2(η2-R−C⋮C−R)]+. Dependence of the Alkenyl Product Stereochemistry on the Basicity of the Nucleophile », Organometallics, vol. 20, no 13, , p. 2736-2750 (DOI 10.1021/om010095t, lire en ligne)
- (en) Yidong Wang, Jin Zhu, Austin C. Durham, Haley Lindberg et Yi-Ming Wang, « α-C–H Functionalization of π-Bonds Using Iron Complexes: Catalytic Hydroxyalkylation of Alkynes and Alkenes », Journal of the American Chemical Society, vol. 141, no 50, , p. 19594-19599 (DOI 10.1021/jacs.9b11716, S2CID 208611984, lire en ligne)
- (en) Bruce M. Foxman, « X-Ray molecular structure of dicarbonyl-η5-cyclopentadienyl-(η2-tetramethylallenyl)iron tetrafluoroborate. A sterically crowded allene complex », Journal of the Chemical Society, Chemical Communications, vol. 1975, no 6, , p. 221-222 (DOI 10.1039/C39750000221, lire en ligne)
- (en) Yidong Wang, Sarah G. Scrivener, Xiao-Dong Zuo, Ruihan Wang, Philip N. Palermo, Ethan Murphy, Austin C. Durham et Yi-Ming Wang, « Iron-Catalyzed Contrasteric Functionalization of Allenic C(sp2)–H Bonds: Synthesis of α-Aminoalkyl 1,1-Disubstituted Allenes », Journal of the American Chemical Society, vol. 143, no 37, , p. 14998-15004 (PMID 34491051, PMCID 8458257, DOI 10.1021/jacs.1c07512, lire en ligne)
- (en) Matthew N. Mattson, Edward J. O'Connor et Paul Helquist, « Cyclopropanation using an iron-containing methylene transfer reagent: 1,1-diphenylcyclopropane », Organic Syntheses, vol. 70, , p. 177 (DOI 10.15227/orgsyn.070.0177, lire en ligne)
- (en) M. D. Johnson, « 31.2 - Mononuclear Iron Compounds with η1-Hydrocarbon Ligands », Comprehensive Organometallic Chemistry, , p. 331-376 (DOI 10.1016/B978-008046518-0.00049-0, lire en ligne)
- (en) Mark Wrighton, « Photochemistry of metal carbonyls », Chemical Reviews, vol. 74, no 4, , p. 401-430 (DOI 10.1021/cr60290a001, lire en ligne)
- (en) Shunichi Fukuzumi, Kei Ohkubo, Mamoru Fujitsuka, Osamu Ito, Marcus C. Teichmann, Emmanuel Maisonhaute et Christian Amatore, « Photochemical Generation of Cyclopentadienyliron Dicarbonyl Anion by a Nicotinamide Adenine Dinucleotide Dimer Analogue », Inorganic Chemistry, vol. 40, no 6, , p. 1213-1219 (DOI 10.1021/ic0009627, lire en ligne)
 Portail de la chimie
Portail de la chimie