Substitution nucléophile
En chimie organique, une réaction de substitution nucléophile est une réaction de substitution au cours de laquelle un groupe nucléophile riche en électrons, noté Nu−, attaque une molécule électrophile ayant un site pauvre en électrons, et remplace un atome ou un groupe d'atomes, appelé groupe partant (noté GP), ou groupe nucléofuge.
Équation générale de ce type de réaction
Les électrons libres (:) du nucléophile Nu− attaquent le substrat R-GP en formant une nouvelle liaison, et entraînant ainsi le départ du groupe partant GP. Parfois, le solvant à lui seul peut provoquer la rupture et laisser le nucléophile réagir. Le nucléophile peut être aussi bien électroniquement neutre, que chargé négativement, tandis que le substrat peut être neutre ou chargé positivement.
Substitution nucléophile d'halogénoalcane
Un exemple de substitution nucléophile est la transformation d'un composé halogéné en alcool (hydrolyse d'un halogénure d'alkyle en milieu basique).
- .

Exemple de substitution d'halogénoalcane



Caractère SN1 ou SN2 d'une réaction
Les réactions de substitution nucléophile doivent être considérées comme un mélange de SN1 et de SN2 selon la nature du groupe partant et de la molécule considérée. Il est donc tout à fait possible qu'une substitution nucléophile conduise à un mélange d'énantiomères ou de diastéréoisomères selon les conditions réactionnelles. Dans l'exemple ci-dessous[1], la réaction décrite inverse proprement la configuration absolue du carbone en α du groupe carboxyle. Cependant, d'après la section “discussion” du même article, si l'on essaye d'activer le groupe hydroxyle en le transformant en sulfonate ou en utilisant une réaction de Mitsunobu, on obtient un mélange d'énantiomères, c'est-à-dire que l'on augmente le caractère SN1 de la substitution.
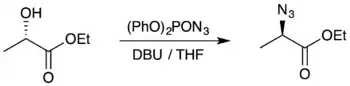
Tableau synthétique des réactions SN1 et SN2
| Type de Réaction | SN1 | SN2 |
|---|---|---|
| Mécanisme |
|
|
| Intermédiaire | Cation, en général un carbocation. | Pas d'intermédiaire (mécanisme à une étape, on peut juste tenter de décrire un "état de transition") |
| Stéréochimie | Mélange racémique, absence de stéréosélectivité. | Inversion de configuration relative ("inversion de Walden"), réaction énantiospécifique |
| Vitesse de réaction | ||
| Influence du radical |
RIII-GP >> RII-GP > RI-GP (stabilisation de l'intermédiaire (par effets inductifs donneurs...)) |
RI-GP > RII-GP >> RIII-GP (déstabilisation de l'état de transition par encombrement stérique) |
| Influence du nucléophile |
La vitesse n'est pas influencée par le nucléophile. | La vitesse augmente avec l'augmentation de :
et diminue quand il est trop volumineux. |
| Influence de la polarité du solvant | Les solvants protiques (eau, méthanol…) favorisent le processus SN1 en facilitant la formation de carbocation par l'établissement de liaisons hydrogène | Les solvants polaires aprotiques (acétone, DMSO…) favorisent le processus SN2 en solvatant le cation associé au nucléophile |
| Influence du nucléofuge |
Plus la liaison est polarisable, plus sa rupture est facile, plus la réaction est rapide. Dans le cas des halogénoalcanes, la vitesse croît de R-F à R-I : R-I > R-Br > R-Cl >> R-F | |


Remarques
- Ce type de réaction est réalisé sous contrôle cinétique : le réactif est à la fois une base[2] de Lewis (caractère thermodynamique), et un nucléophile (caractère cinétique). C'est ici ce dernier caractère qui l'emporte. Peu importe la force du réactif en tant que base, c'est sa force en tant que nucléophile qui joue dans ce type de réaction (c'est strictement l'inverse pour les éliminations).
- Les mécanismes de substitutions nucléophiles monomoléculaires ou bimoléculaires présentés ici et dans leurs propres articles ne sont que des mécanismes limites : aucune réaction chimique réelle ne suit parfaitement l'un de ces deux mécanismes. Il s'agit toujours d'un compromis, plus ou moins proche de l'un ou l'autre. On attribue d'ailleurs souvent à une réaction une sorte de "pourcentage" en mécanismes limites