LKB1
La LKB1 (« Liver kinase B1 » ou kinase hépatique B1) est une enzyme de type kinase, c'est-à-dire capable de phosphoryler d'autres protéines cibles.

Histoire
La protéine LKB1 est codée par le gène LKB1, aussi nommé STK11 pour "Sérine-Thréonine Kinase 11".
LKB1 est une sérine/thréonine kinase présente chez toutes les cellules eucaryotes. Elle est exprimée ubiquitairement et préférentiellement au niveau des villosités épithéliales du petit intestin.
Elle a été identifiée en 1998 comme étant l'un des premiers gène suppresseur de tumeurs[1]. Les mutations de cette protéine ont été associées à la majorité des cas de syndrome de Peutz-Jeghers. Le premier cas clinique de cette maladie a été rapporté en 1896 chez deux jumelles londoniennes, lesquelles présentaient une pigmentation caractéristique de la muqueuse orale. On rapporte par la suite que l'une d'elles est décédée à l'âge de 20 ans d'une pseudo-obstruction intestinal alors que l'autre a développé cancer du sein, succombant à l'âge de 52 ans. Ce n'est qu'en 1954 que le nom de syndrome Peutz-Jeghers a été octroyé à cette maladie, par un clinicien reconnaissant le travail de ses prédécesseurs Peutz et Jaghers qui en avaient clairement identifié les symptômes[2].
Définition
Le gène LKB1/STK11 se retrouve sur le chromosome 19 humain à la position 13.3. La séquence, d’une longueur totale de 23 kb, est constituée de 10 exons dont 9 sont codants. La protéine chez l'homme est composée de 433 acides aminés. La région catalytique (domaine protéine-kinase) se retrouve entre les acides aminés 44 et 309. Fait intéressant, les régions non catalytiques C-Terminale et N-Terminale n'ont pas d'analogie avec d'autres kinases de ce genre et n'ont pas non plus de domaine fonctionnel identifiable[3]. Peutz-Jeghers syndrome protein

Fonctions
Une fois la protéine LKB1 traduite dans le cytoplasme et avant de subir une phosphorylation par différentes kinases, LKB1 est importé dans le noyau afin de former un complexe trimérique avec les pseudo-kinases STRAD et MO25[4]. Le trimère constitue la forme active de LKB1 qui peut alors ajouter un groupement phosphate sur des kinases substrats, afin d'activer celles-ci. La phosphorylation se fait préférentiellement sur la thréonine172 située sur la boucle en "T" (T-loop) de la sous-unité catalytique α de la protéine cible[5].
Les substrats de LKB1 sont plus de 14 kinases[6] dont:
- AMPK α 1 et 2, favorisant la translation des protéines et inhibant la synthèse des acides gras. L'AMPK a été le premier substrat de LKB1 à avoir été identifié. En plus de l'inhibition de la gluconéogenèse par TORC2, l'AMP-kinase influe sur le métabolisme à plusieurs niveaux. À la suite de son activation, l'AMPK favorise les voies catalytiques formant de l'ATP tout en éteignant divers processus en requérant[7].
- BRSK 1 et 2, régulateurs positifs de la polarité neuronale. Au stade embryonnaire, LBK1 par la phosphorylation de BRSK1/2 favorise l'organisation des microtubules au sein des neurones en formation. Une mutation de LKB1 génère des neurones plus courts et dont la migration n'est pas optimale.
- MARK 1 à 4, impliqués dans la stabilité des microtubules et dans la polarité des cellules épithéliales. Grâce à la phosphorylation de MARK 1-4, LKB1 est responsable de l'asymétrie cellulaire apicobasale au niveau de l'épithélium.
- SIK 1 et 2, ayant une action inhibitrice au niveau de la gluconéogenèse dans le foie. De concert avec l'AMP-kinase, SIK1/2 a une action inhibitrice sur le coactivateur TORC2, qui à son tour régule positivement la gluconéogenèse.
- NUAK 1 et 2, régulateurs de l'apoptose via la protéine P53. Dans une lignée de cellules surexprimant LKB1, des expériences en microarray (Puce à ADN) ont révélé que les gènes des protéines associées à la voie de l'apoptose régulée par P53 étaient surexprimés. Ces résultats montrent que LKB1 est positivement impliqué dans l'apoptose et donc dans la suppression des tumeurs.
En plus, LKB1 agit sur la voie de VEGF, le facteur de croissance vasculaire endothélial. Au stade embryonnaire, il a été démontré que LKB1 est essentielle à l'angiogénèse, c'est-à-dire la formation des vaisseaux sanguins chez le fœtus. Des souris déficientes en LKB1 ne parvenaient pas à la fin du stade de gestation en ayant des défauts majeurs au niveau de l'angiogénèse[8]. De même, il facilite l'angiogenèse en réponse à un défaut d'oxygènation (ischémie) tissulaire[9].
Le LKB1 module l'expression de la cavéoline 1 et intervient dans la régulation de l'oxyde nitrique synthase, induisant une vasodilatation[10].
Le LKB1 active également la voie du TGF bêta 1 permettant la prolifération des cellules musculaires lisses vasculaires[11].
Maladies liées
Syndrome de Peutz-Jeghers
Une mutation au niveau du gène LKB1 entraîne la production d'une protéine déficiente, ayant une activité diminuée ou abolie. Dans 80 % des cas de syndrome de Peutz-Jeghers, une mutation de LKB1 est constatée[12]. À ce jour, au moins 75 mutations de la protéine ont été identifiées, dont la plupart affectent la partie catalytique de la protéine, ayant des conséquences plus graves que les mutations au niveau non-catalytique. Le syndrome de peutz-Jeghers est une maladie autosomale dominante et héréditaire, touchant seulement une personne sur 100 000. Il s'agit en fait d'un syndrome de prédisposition au cancer. Ses symptômes mineurs comportent des défauts de pigmentation autour des lèvres, au niveau du visage est des organes génitaux. Dans les cas plus sévères, le tractus gastro-intestinal est parsemé de polypes bénins. Dans 93 % des cas, ces polypes vont devenir des adénomes malins et éventuellement métastaser au niveau de la rate, du foie, des ovaires et des seins. 48 % des patients vont succomber à un cancer avant l'âge de 57 ans.
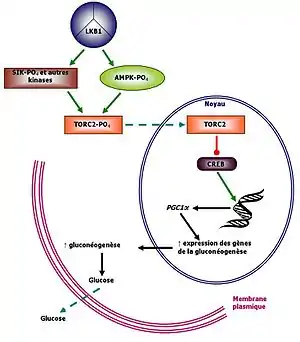
Diabète de type 2
Comme montré sur le schéma ci-contre, LKB1 agit comme régulateur de l'homéostasie du glucose dans le foie. Il est répresseur de la gluconéogenèse en activant TORC2(Transducer of Regulated CREB Activity) via la phosphorylation de l'AMPK. TORC a un effet inhibiteur sur le facteur de transcription CREB (cAMP-Response-Element-Binding). CREB régule la transcription d'une panoplie de gènes, dont ceux impliqués dans le métabolisme du glucose. En cas d'une mutation de LKB1, cette cascade régulatrice est perturbée, menant à une augmentation de l'expression des gènes de la gluconéogenèse. Il y a ensuite relargage de glucose dans le système, créant une hyperglycémie et le sujet atteint a des symptômes de diabète de type 2[13].
L'incidence du diabète de type II relié à une déficience en LKB1 n'a pas encore été démontrée. Cependant, une étude rétrospective a démontré que les personnes traitées avec la metformine étaient moins à risque d'avoir des cancers. Par un mécanisme inconnu, la metformine phosphoryle l'AMPK au même titre que LKB1. Des études sont actuellement en cours afin d'étudier une voir de traitement possible du diabète de type II chez des patients présentant une LKB1 mutée.
Cancers
De tous les cancers pulmonaires, l'adénocarcinome est en cause dans 40 % des cas. De ceux-ci, 30 % sont dus à des dérèglements ou à des mutations spontanées de LKB1. Ceci représente chaque année aux États-Unis 12 500 décès. Étonnamment, les Japonais semblent moins sujets à des mutations de LKB1 que les caucasiens, en ce qui a trait à l'adénocarcinome pulmonaire[14].
Les mutations du gènes facilites également la survenue du cancer du col utérin[15] ou de l'endomètre[16].
Notes et références
- Ylikorkala A, Avizienyte E, Tomlinson I P, Tiainen M, Roth S, Loukola A, Hemminki A, Johansson M, Sistonen P, Markie D, Neale K, Phillips R, Zauber P, Twama T, Sampson J, Jarvinen H, Makela T P, Aaltonen L A (1999) "Mutations and impaired function of LKB1 in familial and non-familial Peutz–Jeghers syndrome and a sporadic testicular cancer" Human Molecular Genetics 8:45–51.
- McGarrity T J, Amos C (2006) "Peutz-Jeghers syndrome: clinicopathology and molecular alterations! Cellular and Molecular Life Sciences 63:2135–2144
- Boudeau J, Sapkota G, Alessi D R (2003) "LKB1, a protein kinase regulating cell proliferation and polarity" FEBS Letters 546:159-165
- Boudeau J, Scott J W, Resta N, Deak M, Kieloch A, Komander D, Hardie D G, Prescott A R, van Aalten D M F, Alessi1 D R (2004) "Analysis of the LKB1-STRAD-MO25 complex" Journal Of Cell Science 117:6365-6375
- Katajisto P, Vallenius T, Vaahtomeri K, Ekman N, Udd L, Tiainen M, Mäkelä T (2007) "The LKB1 tumor suppressor kinase in human disease" Biochimica et Biochimica Acta 1775:63-75
- Alessi DR, Sakamoto K, Bayascas JR, LKB1-dependent signaling pathways, Annu Rev Biochem, 2006;75:137–163
- Hardie D G (2005) "New roles for the LKB1→AMPK pathway" Current Opinion In Cell Biology 17:167-173
- Ylikorkala A, Rossi D J, KorsisaariN, Luukko K, Alitalo K, Henkemeyer M, Mäkelä T P (2001)"Vascular Abnormalities and Deregulation of VEGF in Lkb1-Deficient Mice" Science 293(5533):1323-1326
- Ohashi K, Ouchi N, Higuchi A, Shaw RJ, Walsh K, LKB1 deficiency in Tie2-Cre-expressing cells impairs ischemia-induced angiogenesis, J Biol Chem, 2010;285:22291–22298
- Zhang W, Wang Q, Wu Y et al. Endothelial cell–specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo, Circulation, 2014;129:1428-1439
- Londesborough A, Vaahtomeri K, Tiainen M, Katajisto P, Ekman N, Vallenius T, Mäkelä TP, LKB1 in endothelial cells is required for angiogenesis and TGFbeta-mediated vascular smooth muscle cell recruitment, Development, 2008;135:2331–2338
- Bignell G R, Barfoot R, Seal S, Collins N, Warren W, Stratton M R (1998) "Low Frequency of Somatic Mutations in the LKB1/Peutz-Jeghers Syndrome Gene in Sporadic Breast Cancer" Cancer Research 58:1384-1386
- Carling D (2006) "LKB1: a seet side to Peutz-Jeghers syndrome?" TRENDS in Molecular Medicine 12(4):144-147
- Onozato R, Kosaka T, Achiwa H, Kuwano H, Takahashi T, Yatabe Y, Mitsudomi T (2007) "LKB1 gene mutations in Japanese lung cancer patients" Cancer Science 98(11):1747-1751
- Wingo SN, Gallardo TD, Akbay EA et al. Somatic LKB1 mutations promote cervical cancer progression, PLoS One, 2009;4:e5137
- Contreras CM, Gurumurthy S, Haynie JM et al. Loss of Lkb1 provokes highly invasive endometrial adenocarcinomas, Cancer Res, 2008;68:759–766
 Portail de la biologie
Portail de la biologie  Portail de la médecine
Portail de la médecine