Lissencéphalie
La lissencéphalie désigne un ensemble de maladies qui ont en commun la disparition de l’aspect habituel du cortex cérébral.
| Spécialité | Génétique médicale et neurologie |
|---|
| CISP-2 | N85 |
|---|---|
| CIM-10 | Q04.3 |
| OMIM | 615191, 300067, 607432, 614019 et 300215 611603, 615191, 300067, 607432, 614019 et 300215 |
| DiseasesDB | 29492 |
| MeSH | D054082 |
| GeneReviews | et NBK5189 |
![]() Mise en garde médicale
Mise en garde médicale
Le cortex cérébral est habituellement traversé par des sillons lui donnant un aspect de « champ labouré ». Certains de ces sillons ont un aspect plus important et sont appelés des scissures. Le processus de formation des sillons et des scissures s’appelle la gyration.
La lissencéphalie est donc une anomalie de la gyration : soit il n’existe plus du tout de sillon et on parle d’agyrie, soit il existe encore quelques sillons on parle de pachygyrie.
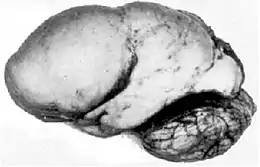
En cas de lissencéphalie, on ne voit plus de sillon et le cerveau apparaît lisse, ce qui explique le terme de lissencéphalie, l'encéphale désignant le cerveau.
En plus de l’aspect macroscopique de la maladie, il s’y ajoute une désorganisation microscopique des couches de neurones du cortex.
Étiologie
Quatre phénomènes peuvent être à l’origine d’une anomalie de la gyration :
- une anomalie génétique venant de l’un ou des deux parents (mutation du chromosome X) ;
- une anomalie génétique apparaissant soudainement dans un gène important pour une bonne formation cérébrale ;
- une infection, surtout virale, au cours de la grossesse ;
- une diminution de la vascularisation cérébrale fœtale.
Incidence
Un sur 100,000 naissances.
Diagnostic
Le diagnostic anténatal est possible et dépend du type de la lissencéphalie. Par contre, le diagnostic précis de lissencéphalie est indispensable afin de conseiller les parents. L'imagerie à résonance magnétique est indispensable pour établir un diagnostic précis et pour conseiller les parents. Si les parents décident une interruption médicale de grossesse, un examen anatomo-pathologique du cerveau est indispensable pour établir un diagnostic précis.
Conséquence d’un trouble de la gyration
- Certains types particulièrement sévères de lissencéphalie entraînent le décès de l’enfant en quelques mois.
- Les troubles de la gyration s’accompagnent d’un retard mental dont la gravité dépend du type de la lissencéphalie et de son importance. Parfois seule une partie du cerveau est atteinte.
- Des crises d'épilepsie surviennent chez un grand nombre d’enfants. Un traitement antiépileptique permet de contrôler ces crises.
- Des troubles de l’alimentation sont aussi présents nécessitant des interventions chirurgicales ou des prothèses pour assurer une alimentation normale.
Génétique
On connaît cinq gènes dont le dysfonctionnement provoque une lissencéphalie chez l’humain[1]. Le type de lissencéphalie observée est sensiblement différent dans les cinq cas que nous allons évoquer successivement ici.
LIS1 et DCX
Les cas de lissencéphalie classique les plus fréquemment rencontrés sont causés par des mutations dans le gène LIS1[2] ou le gène DCX[3],[4]. La différence principale qui existe entre les deux types de lisencéphalie est d’ordre topographique : alors que des mutations dans LIS1 provoquent des lésions principalement dans les régions postérieures du cortex cérébral, les mutations dans DCX provoquent des lésions antérieures. Des études récentes ont montré que ces deux protéines sont capables d’interagir avec les microtubules et d’intervenir au niveau des protéines du centrosome (en particulier la gamma-tubuline et la dynéine). La protéine doublecortine fait partie de la famille des MAP (« microtubule associated proteins ») qui posséderait un rôle dans la stabilisation de cette structure cellulaire essentielle aux mécanismes de migration. On sait par ailleurs aujourd’hui que les protéines LIS1 et DCX sont capables d’interagir in vitro. Comment une anomalie dans ces protéines conduit au phénotype cérébral observé n’est pas encore compris (revue dans Gupta et al. 2002[source insuffisante]).
RELN
Les cas de lissencéphalie dus aux mutations dans le gène RELN[5], codant la reeline, perturbent moins sévèrement l’organisation spatiale des couches corticales, et on parle plutôt de pachygyrie que d’agyrie en ce qui concerne le cerveau de ces patients. De plus, ils présentent une atteinte cérébelleuse majeure qui est caractéristique si elle est comparée aux patients présentant une mutation dans le gène LIS1 ou DCX. Le gène RELN a été isolé chez l’humain après que le gène homologue chez la souris ait été extensivement caractérisé (D’Arcangelo et al., 1995). Le gène murin reelin est le gène qui est défectueux dans le mutant reeler. Les animaux mutants ont un phénotype qui consiste en une anomalie de la posture et des défauts de lamination corticale (dans le néocortex et le cervelet). La reeline, protéine codée par le gène RELN, est une grosse protéine sécrétée exprimée surtout dans la zone marginale du néocortex ainsi que dans le cervelet. Cette protéine est un ligand pour le récepteur des lipoprotéines de très faible densité (VLDLR) et le récepteur de l’apolipoprotéine E (ApoER2) (D’Arcangelo et al., 1999). Des études génétiques ont montré que si ces deux récepteurs étaient anormaux, le phénotype obtenu est semblable à celui du mutant Reeler[6]. Des études génétiques ont par ailleurs permis de montrer qu’un autre mutant murin, le mutant scrambler, est dû à une mutation dans le gène Dab-1 et que ce gène code une protéine cytoplasmique capable de reconnaître spécifiquement les parties intracytoplasmiques des récepteurs ApoER2 et VLDLR. Grâce à l’étude des mutants animaux spontanés, un mécanisme original a donc été identifié reliant un ligand (RELN) à ses récepteurs spécifiques (VLDLR et ApoER2). On sait aujourd’hui que ces deux récepteurs sont capables d’induire la phosphorylation de Dab-1 en réponse à la liaison de RELN ou à un clivage protéolytique médié par RELN (dont on pense qu’elle pourrait également posséder une activité protéase). Si l’on empêche artificiellement Dab-1 d’être phosphorylée in vivo, on obtient des animaux qui présentent des anomalies dans le positionnement des neurones. Les études biochimiques et génétiques réalisées dans ce cas montrent clairement l’intérêt essentiel de l’étude des perturbations naturelles de la migration neuronale pour mettre en évidence les voies de signalisation qui sont utilisées dans les processus normaux de migration[7].
ARX
Le gène de lissencéphalie le plus récemment identifié est ARX (Stromme et al., 2002). Des mutations dans le gène ARX provoquent une lissencéphalie à prédominance frontale d’un type relativement différent de celui observé dans le cas de mutations dans DCX, LIS1 ou RELN (en particulier en ce qui concerne l’épaisseur du cortex). On connaît des mutations dans ce gène qui provoquent également, en association avec la lissencéphalie, une agénésie du corps calleux et/ou une ambigüité sexuelle. Un très large spectre phénotypique est donc associé aux mutations dans ARX sans que l’on puisse l’expliquer à l’heure actuelle. ARX est un gène à homéoboite exprimée principalement dans le cerveau adulte et fœtal ainsi que dans le muscle squelettique. Chez la souris, ce gène est surtout exprimé dans le cortex frontal, en bon accord avec le phénotype à prédominance frontale qui est observé chez l’humain. La protéine ARX est supposée être un répresseur de la transcription bien que cela reste à démontrer. Alors que les gènes LIS1 et RELN possèdent une localisation autosomique (sur les chromosomes 17p13 et 7q22 respectivement), les gènes DCX et ARX sont tous deux localisés sur le chromosome X humain. Cela signifie donc dans ces deux derniers cas que le mode d’hérédité est directement influencé par le sexe. Les femmes pourront être des conductrices saines ou bien présenter un phénotype atténué par rapport aux hommes porteurs des mêmes mutations (phénotype de d’hétérotopie subcorticale en bande (ou SBH) pour les conductrices d’une mutation dans le gène DCX). La connaissance du défaut moléculaire initial dans le cadre d’un tableau clinique de lissencéphalie a donc un impact direct sur le type de conseil génétique qui sera donné aux familles concernées.
TUBA3 / TUBA1A
Le gène de lissencéphalie le plus récemment identifié est le gène de la tubuline alpha-1 appelé TUBA3 ou TUBA1A (Keays et al., 2007). Ce gène a été isolé sur la base de l'existence d'un modèle murin présentant une lamination anormale de l'hippocampe. Les mutations dans ce gène sont transmises selon un mode d'hérédité autosomique dominante. Outre la lissencéphalie, les patients présentent des anomalies au niveau du corps calleux et du tronc cérébral (Poirier et al., 2007).
Différents types de lissencéphalie
Il existe plusieurs types de lissencéphalie dont la distinction est importante en raison de leur mode de transmission différente.
Lissencéphalie classique
- Lissencéphalie classique ou lissencéphalie liée au chromosome 17 ou syndrome de Miller-Dieker
- Lissencéphalie due à la mutation du gène double cortine
- Lissencéphalie inexpliquée
Lissencéphalie avec microcéphalie
Lissencéphalie pavimenteuse
Dans ce type de lissencéphalie, qui est le résultat d'une désorganisation globale de la formation du cortex, la surface cérébrale est très irrégulière, ressemblant au pavage d'une rue, d'où le nom de ce type de lissencéphalie.
Notes et références
- (en) Renzo Guerrini et Carla Marini, « Genetic malformations of cortical development », Exp Brain Res, vol. 173, no 2, , p. 322-33. (PMID 16724181, DOI 10.1007/s00221-006-0501-z).
- (en) Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH, « Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13 », JAMA, vol. 270, no 23, , p. 2838-42. (PMID 7907669, DOI 10.1001/jama.1993.03510230076039).
- (en) Pilz DT, Matsumoto N, Minnerath S, Mills P, Ross ME et al., « LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation », Hum Mol Genet, vol. 7, no 13, , p. 2029-37. (PMID 9817918, DOI 10.1093/hmg/7.13.2029, lire en ligne [html]).
- (en) des Portes V, Pinard JM, Billuart P, Vinet MC, Chelly J et al., « A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome », Cell, vol. 92, no 1, , p. 51-61. (PMID 9489699, DOI 10.1016/S0092-8674(00)80898-3, lire en ligne [PDF]).
- (en) Hong SE, Shugart YY, Huang DT, Shahwan SA, Walsh CA et al., « Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations », Nat Genet, vol. 26, no 1, , p. 93-6. (PMID 10973257, DOI 10.1038/79246).
- (en) Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J, « Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2 », Cell, vol. 97, no 6, , p. 689-701. (PMID 10380922, DOI 10.1016/S0092-8674(00)80782-5, lire en ligne [PDF]).
- (en) Tissir F, Goffinet AM, « Reelin and brain development », Nat Rev Neurosci, vol. 4, no 6, , p. 496-505. (PMID 12778121, DOI 10.1038/nrn1113).
Liens externes
- Page spécifique sur Orphanet
- Site en anglais Incontournable pour les maladies génétiques
- Site en anglais spécialisé dans la lissencéphalie
- NIH
- Lissencephaly Launch Pad (support)
 Portail de la médecine
Portail de la médecine