Maladie de Behçet
La maladie de Behçet est une vascularite systémique, maladie liée à l'inflammation des vaisseaux sanguins de l'organisme.
| Causes | Système immunitaire |
|---|---|
| Symptômes | Aphte, ulcération de la vulve (d), érythème noueux, vascularite pustuleuse (d), uvéite, vascularite rétinienne (en), hypopion (en), douleur abdominale, nausée, diarrhée, hémoptysie, pleurésie, toux, fièvre, arthrite, méningoencéphalite (en), méningite aseptique (en), péricardite, pathergie (en), acné, chronic widespread pain (d) et inflammation |
| Traitement | Corticoïde, Inhibiteur du TNF, Interferon alpha 2 (d), azathioprine et thérapie par immunoglobulines |
|---|---|
| Médicament | Thalidomide, aprémilast, ciclosporine, étanercept, adalimumab, (S)-(−)-colchicine et infliximab |
| Spécialité | Immunologie et rhumatologie |
| CIM-10 | M35.2 |
|---|---|
| CIM-9 | 279.4 |
| OMIM | 109650 |
| DiseasesDB | 1285 |
| eMedicine |
1006358 ped/219 derm/49 oph/425 |
| MeSH | D001528 |
![]() Mise en garde médicale
Mise en garde médicale
Elle se déclare essentiellement chez l'adulte jeune, et se manifeste le plus souvent par des symptômes cutanéo-muqueux (aphtose), articulaires (douleur) et ophtalmiques (uvéite). Plus rarement les lésions peuvent atteindre le système nerveux (méningite, thrombose veineuse cérébrale), digestif (douleur, saignement) et cardio-vasculaire (thrombose, anévrisme).
C'est une maladie chronique qui évolue par poussées d'intensités variables. À terme le pronostic est principalement lié au risque de séquelles en cas de poussées sévères, notamment oculaires et neurologiques.
Plus fréquente au Moyen-Orient et en Asie de l'Est, c'est une maladie rare en Europe et en Amérique du Nord.
Comme la plupart des vascularites, le traitement, non curateur, repose essentiellement sur les immunosuppresseurs et a pour but de réduire la fréquence et la sévérité des poussées et prévenir leurs complications.
Historique
Initialement, la maladie de Behçet a été décrite sous forme d'une triade : aphtose buccale, aphtose génitale et uvéite. Le dermatologue turc Hulusi Behçet a décrit cette maladie en 1937, mais on trouve dans le traité Épidémies III d'Hippocrate une description compatible[1],[2].
Cause
Elle est inconnue. Elle associe probablement un phénomène inflammatoire avec une autoimmunité, faisant intervenir l'interleukine 21 et différents types de lymphocytes[3]. Il existe également une activation des polynucléaires neutrophiles[4].
Épidémiologie
La maladie de Behçet s'observe essentiellement dans les pays du bassin méditerranéen, au Moyen et en Extrême-Orient[5]. En France, on ne compte pas plus de 100 cas en 2012, soit une prévalence inférieure à 6 par 100 000, ce qui la rend extrêmement rare. En Turquie, sa prévalence est de 80 à 300 par 100 000 personnes selon les régions. Au Japon, 10 personnes sur 100 000 en sont atteintes.
La connotation génétique est probable du fait d'un lien fort avec l'antigène HLA B5101 (antigène associé à la maladie de Behçet dans 50 à 70 % des cas), mais les cas familiaux sont rares, ce qui est contradictoire. La prédominance masculine qui paraissait nette est aujourd'hui controversée. Elle débute chez l'adulte jeune (18 à 40 ans), rarement chez l'enfant et exceptionnellement après 50 ans.
Signes cliniques
Les signes sont souvent non spécifiques, ce qui explique que le diagnostic peut être retardé de plusieurs années de manière courante[6]. L'atteinte peut différer suivant la population ou le sexe.
Signes cutanés
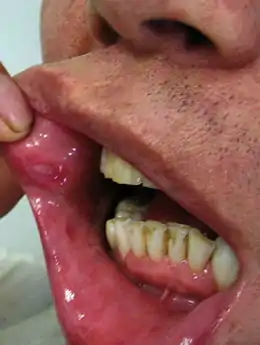
- Aphtes buccaux : ulcérations à bords nets érythémateux (rouges), surélevés (1-3 cm de diamètre), douloureux, isolées ou en groupes (grappes), évoluant par poussées d'intensité variable[7]. Cette atteinte est quasi constante dans la maladie[8]. Le caractère récidivant est un indice en faveur d'un Behçet[9].
- Aphtes génitaux, inconstants (dans 50 % des cas[7] à 80 % des cas[8]), très évocateurs du Behçet, pouvant laisser des cicatrices qu'il faut savoir interpréter; de même pour de rares affections du scrotum[10].
- Pseudo-folliculites cutanées : elles sont fréquentes (60 % des cas). Elles siègent au dos, au visage, aux membres inférieurs, aux fesses et aux bourses. Ce sont des papules qui se recouvrent en deux ou trois jours d’une vésicule qui devient une pustule, puis d’une croûte qui se détache sans laisser de trace. Le contenu des vésicules est stérile. Cette lésion est non centrée par un poil.
- Hyper-réactivité aux traumatismes, voire authentique érythème noueux dans un peu moins d'un cas sur deux[8].
- Phénomène pathergique cutané : il s’agit d’une hypersensibilité aux points cutanés. La piqûre se fait avec une aiguille stérile G20 à la face antérieure de l’avant bras en sous cutané et/ou en intraveineux et la lecture se fait après 48 heures. Ce test reproduit la pseudo-folliculite nécrotique qui apparaît spontanément. La papule n’a de valeur que si le diamètre est supérieur à 2 mm[11]. Ce test n'est cependant positif que dans certaines populations[12].
{kind=link}
Signes oculaires
Les atteintes oculaires conditionnent le pronostic fonctionnel par le risque de cécité qu'elles impliquent. L'uvéite antérieure à hypopion est caractéristique. L'uvéite postérieure est plus dissimulée, mais de pronostic parfois sévère. Toutes les parties de l'œil peuvent être atteintes. En cas d'atteinte oculaire, le pronostic visuel est médiocre malgré le traitement, avec l'évolution vers une cécité dans un quart des cas[13]. Ce pronostic est cependant bien amélioré avec l'administration d'un anticorps chimérique anti TNF-Alpha : l'infliximab[14].
Signes vasculaires : vascularites
Il s'agit essentiellement de thrombose de gros vaisseaux (veines ou artères) survenant sur une inflammation de leur paroi[15].
- Atteintes veineuses avec thrombophlébites superficielles, profondes, ou viscérales.
- Atteintes artérielles avec thromboses, anévrismes, de n'importe quelle artère.
- Manifestations cardiaques touchant l'endocarde, le myocarde, le péricarde ou les coronaires[16].
Signes articulaires
Ils sont fréquents, entre 50 et 60 %[11]. Les principales caractéristiques sont[11] :
- la prédilection pour les grosses articulations des membres inférieurs notamment les genoux et les hanches ;
- la guérison sans séquelles ;
- leur caractère récidivant ;
- l’absence de déformation et de destruction.
L'atteinte articulaire revêt plusieurs aspects[11] :
- mono ou oligoarthrite aiguë ;
- monoarthrite chronique ;
- polyarthrite bilatérale plus ou moins symétrique simulant une polyarthrite rhumatoïde ;
- l’atteinte érosive est rare.
Signes neurologiques
Des manifestations neurologiques sporadiques (neuro-Behçet) sont fréquentes (plus de 20 %), elles apparaissent souvent entre 1 et 10 ans après les manifestations initiales : céphalées, signes pyramidaux avec hémiparésie, changements de comportement (symptômes schizophréniformes, apparition de troubles addictifs...) et dysfonctionnements sphinctériens. Elles peuvent être graves, dominées par la méningo-encéphalite. Peuvent être présentes des myélites inflammatoires qui peuvent déboucher sur un diagnostic différentiel d'une poussée de sclérose en plaques (SEP). Des symptômes psychiatriques ont été décrits dans la moitié des cas de neuro-Behçet. Le délai moyen d'apparition des troubles psychiques est de 26,2 mois. Les troubles psychiatriques sont traités par des neuroleptiques, des anxiolytiques et des antidépresseurs. L'évolution de l'atteinte psychique est favorable dans 62 % des cas. Les troubles psychiques au cours de la maladie de Behçet sont d'un grand polymorphisme clinique et surviennent surtout chez les patients avec atteinte neurologique, nécessitant une prise en charge multidisciplinaire.
Signes gastro-intestinaux
Ils sont plus rares et peu spécifiques. Quelques cas de pancréatite ont été décrits. Un diagnostic erroné de maladie de Crohn est fort probable en raison des lésions intestinales très semblables dans la maladie de Behçet. Plusieurs cas de diagnostic erroné prouvent qu'il faut user de vigilance lors du diagnostic différentiel.
Des lésions aphtoïdes ou ulcéreuses peuvent affecter le tube digestif, surtout la jonction iléo-cæcale et le côlon ascendant, entraînant potentiellement hémorragies et perforations.
Signes respiratoires
Des infiltrats pulmonaires et des pleurésies sont possibles. Les possibilités à éliminer sont l'embolie pulmonaire et la vascularite des artères pulmonaires, voire des anévrismes des artères pulmonaires.
Signes rénaux
Il est parfois relevé un taux élevé de protéines dans les urines, associé à des phénomènes de lithiases urinaires (calculs urinaires entraînant des crises de coliques néphrétiques). La glomérulopathie subaiguë reste rare.
Examens complémentaires
La biologie montre une hyperleucocytose à prédominance de polynucléaires neutrophiles (très peu spécifique). Il existe un syndrome inflammatoire variable. Le groupage HLA n'a qu'un intérêt épidémiologique et non pas diagnostique.
La biopsie des lésions cutanées montre un infiltrat de polynucléaires neutrophiles[17].
Critères diagnostiques
Les critères de classification internationaux, définis par la présentation clinique, sont sensibles et spécifiques[18] :
- critère obligatoire : présence d'aphtes buccaux récurrents, au moins trois fois en 12 mois ;
- critères secondaires (au moins deux) :
- ulcération génitale récurrente,
- lésions oculaires,
- lésions cutanées ou pathergie.
Nouvelle classification (critères internationaux 2013). Le diagnostic est retenu si le score est égal ou supérieur à 4:
- Atteinte ophtalmologique: 2 points
- Aphtose génitale: 2 points
- Aphtose buccale; 2 points
- Manifestations cutanées; 1 point
- Manifestations neurologiques; 1 point
- Manifestations vasculaires; 1 point
- Test de pathergie; 1 point
Pour le neuro-Behçet, la ponction lombaire est obligatoire, l'IRM peut révéler des lésions inflammatoires du tronc cérébral et des hémisphères cérébraux (hypersignaux en séquence T2).
Diagnostics différentiels
On peut citer[7] :
- uvéite infectieuse
- polychondrite récidivante
- sarcoïdose
- syndrome des antiphospholipides
- artérite de Takayasu
- maladie de Crohn
- sclérose en plaques.
Traitement
Sa prise en charge a fait l'objet de la publication de recommandations. Celle de l'European League against rheumatism datent de 2008[19].
Il n'existe pas de traitement curatif ; la prise en charge est donc basée sur le soulagement des symptômes (traitement symptomatique) et la prévention des complications.
La colchicine a un effet sur les aphtes et les affections articulaires (traitement de première intention des formes cutanéomuqueuses simples )[7],[20]. Elle est également efficace sur les atteintes cutanées[21].
Les stéroïdes anti-inflammatoires (corticoïdes) sont la base du traitement des formes cutanées résistantes à la colchicine ou plus graves. Ils ne sont pas efficaces dans les lésions muqueuses[22]. Néanmoins la corticodépendance et des rechutes peuvent se produire à l'arrêt du traitement[7],[20]. Leur administration peut être en bolus intraveineux (doses de charge) ou en traitement continu per os.
Les immunosuppresseurs (en général azathioprine, cyclophosphamide, méthotrexate, mycophénolate mofétil) peuvent aussi être utilisés en administration concomitante, mais leur action est lente[7],[20]. Leur utilisation permet de diminuer les corticoïdes sans présenter une rechute de la maladie, mais ils augmentent le risque d'infection[20].
L’interféron alpha peut être utilisé dans certaines formes oculaires sévères[14], en injections sous-cutanées[7],[20]. Les anticorps anti-TNF alpha chimériques sont utilisés dans les formes sévères de la maladie ou les formes résistantes aux traitements précédents[20]. Les timbres de nicotine ont eu un effet bénéfique sur les signes cutanés (uniquement) dans une très courte série (cinq cas) publiée en 2010[23].
Un soutien psychologique peut aider à mieux accepter la maladie et la vivre au mieux au quotidien.
Notes et références
- Mirko D. Grmek, Les maladies à l'aube de la civilisation occidentale, Paris, Payot, , p. 217.
- Hippocrate (éd. Littré), Œuvres complètes, vol. 3, Paris, Baillière, (lire en ligne), p. 84.
- (en) Pineton de Chambrun M, Wechsler B, Geri G, Cacoub P, Saadoun D, « New insights into the pathogenesis of Behçet’s disease », Autoimmun Rev. 2012;11:687-698
- Eksioglu-Demiralp E, Direskeneli H, Kibaroglu A, Yavuz S, Ergun T, Akoglu T, Neutrophil activation in Behçet’s disease, Clin Exp Rheumatol, 2001;19(5 Suppl 24):S19–S24
- (en) Davatchi F, Shahram F, Chams-Davatchi C et al. « Behcet’s disease: from East to West » Clin Rheumatol. 2010;29:823-833
- (en) Ideguchi H, Suda A, Takeno M, Ueda A, Ohno S, Ishigatsubo Y, « Behçet disease: evolution of clinical manifestations », Medicine (Baltimore), 2011;90:125-132
- David Saadoun, « Maladie de Behçet », sur www.orpha.net Orphanet, (consulté le )
- (en) Alpsoy E, Donmez L, Onder M et al. « Clinical features and natural course of Behçet’s disease in 661 cases: a multicentre study », Br J Dermatol. 2007;157:901-906
- (en) Bang D, Hur W, Lee ES, Lee S, « Prognosis and clinical relevance of recurrent oral ulceration in Behçet’s disease », J Dermatol. 1995;22:926-929
- Bouomrani S (2015) Vascularite gangréneuse juvénile du scrotum au cours de la maladie de Behçet. Research fr.
- Hasna Hassikou, « La maladie de Behçet », sur www.rhumato.info, (consulté le )
- (en) Yazici H, Chamberlain MA, Tüzün Y, Yurdakul S, Müftüoglu A, « A comparative study of the pathergy reaction among Turkish and British patients with Behçet’s disease » Ann Rheum Dis. 1984;43:74-75
- (en) Kitaichi N, Miyazaki A, Iwata D, Ohno S, Stanford MR, Chams H, « Ocular features of Behcet’s disease: an international collaborative study » Br J Ophthalmol. 2007;91:1579-1582
- (en) Keino H, Okada AA, Watanabe T, Taki W, « Long-term efficacy of infliximab on background vascular leakage in patients with Behçet's disease », Eye (Lond), vol. 28, no 9, , p. 1100-6. (PMID 24946845, DOI 10.1038/eye.2014.138)
- Calamia KT, Schirmer M, Melikoglu M, Major vessel involvement in Behçet’s disease: an update, Curr Opin Rheumatol, 2011;23:24–31
- (en) Miloslavsky E, Unizony S, « The heart in vasculitis » Rheum Dis Clin North Am. 2014;40:11-26
- (en) Demirkesen C, Tüzüner N, Mat C et al. « Clinicopathologic evaluation of nodular cutaneous lesions of Behçet syndrome » Am J Clin Pathol. 2001;116:341-346
- Polycopié du Collège des Enseignants en Dermatologie de France (CEDEF), 2010-2011 (lire en ligne), Item 343 - Page 8
- (en) Hatemi G, Silman A, Bang D et al. « EULAR recommendations for the management of Behçet disease » Ann Rheum Dis. 2008;67:1656-1662
- David Saadoun (service de médecine interne et d’immunologie clinique, centre national de référence des maladies autoimmunes et systémiques rares, DHU I2B, inflammation, immunopathologie, biothérapie, UPMC, Paris VI, CHU Pitié-Salpêtrière, Paris), « Behçet (maladie de) | SNFMI », sur www.snfmi.org Société nationale française de médecine interne, (consulté le )
- (en) Davatchi F, Sadeghi Abdollahi B, Tehrani Banihashemi A et al. « Colchicine versus placebo in Behçet’s disease: randomized, double-blind, controlled crossover trial » Mod Rheumatol. 2009;19:542-549
- (en) Mat C, Yurdakul S, Uysal S et al. « A double-blind trial of depot corticosteroids in Behçet’s syndrome » Rheumatology (Oxford) 2006;45:348-352
- (en) Ciancio G et al. « Nicotine-patch therapy on mucocutaneous lesions of Behçet’s disease: a case series » Rheumatology 2010;49:501-504.
 Portail de la médecine
Portail de la médecine