Neurofibromatose de type I
La neurofibromatose de type I est aussi appelée maladie de Recklinghausen du nom du médecin allemand, Friedrich Daniel von Recklinghausen, qui le premier a décrit cette maladie en 1881. C'est une maladie monogénique neurodéveloppementale, caractérisée par des symptômes multisystémiques incluant une augmentation du risque de déficit cognitif (50-70%)[1] et une prédisposition à la formation de tumeurs[2]. Elle est une des plus fréquentes maladies génétiques à transmission autosomique dominante. Le gène NF1 est un des gènes dont le taux de mutation spontanée est un des plus importants chez l'homme[réf. nécessaire] : environ la moitié des personnes atteintes par cette maladie est le résultat d'une mutation de novo.
Cet article porte sur la neurofibromatose de type I qui n'a aucun rapport, clinique ou génétique, avec la neurofibromatose de type II.

| Spécialité | Génétique médicale et neurologie |
|---|
| CIM-10 | Q85.0 (ILDS Q85.010) |
|---|---|
| CIM-9 | 237.71 |
| OMIM | 162200 |
| DiseasesDB | 8937 |
| MedlinePlus | 000847 |
| eMedicine |
1112001 neuro/248 oph/338 radio/474 |
| MeSH | D009456 |
| GeneReviews | et NBK47312 |
![]() Mise en garde médicale
Mise en garde médicale
| Neurofibromatose de type I | |
| Référence MIM | 162200 |
|---|---|
| Transmission | Dominante |
| Chromosome | 17q11.2 |
| Gène | NF1 |
| Empreinte parentale | Non |
| Mutation | Plus de 500 connues |
| Mutation de novo | 50 % |
| Anticipation | Non |
| Porteur sain | sans objet |
| Incidence | 1⁄4,000 |
| Prévalence | 1⁄5,000 |
| Pénétrance | 100 % |
| Maladie génétiquement liée | Syndrome de Watson |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
Épidémiologie
Son incidence est estimée à 1⁄4000 et sa prévalence à 1⁄5000[3].
Mécanisme
Le gène NF1 code une protéine, la neurofibromine, formée de plus de 2 800 acides aminés et qui ferait partie de la famille des enzymes hydrolysant la GTP (Guanosine triphosphate)[4]. Le gène NF1 est localisé sur le chromosome 17 en position 17q11.2.
Le gène NF1 est un des gènes dont le taux de mutation spontanée est un des plus importants chez l'homme[réf. souhaitée] : environ la moitié des personnes atteintes par cette maladie est le résultat d'une mutation de novo[5].
Symptômes
Taches « café au lait »
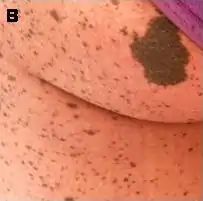
Les taches « café au lait » sont des taches de couleur variant du beige clair au marron foncé et dont la taille varie pour un adulte de quelques millimètres à plusieurs centimètres.
Ces taches sont très fréquentes dans la population saine (plus de 10 % de la population en possède une ou deux), mais dans le cas de la NF-1 leur nombre est de 6 au minimum, certains malades en ayant parfois plusieurs dizaines. Ces taches peuvent disparaître à l'âge adulte.
Elles apparaissent souvent dès la naissance. Pour être comptabilisées, elle doivent mesurer au minimum 5 mm chez un enfant et 1,5 cm chez un adulte et être au nombre de six minimum.
Neurofibromes
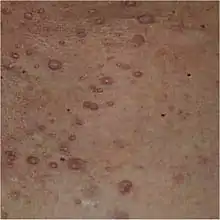
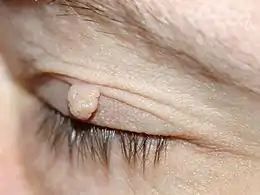
Les neurofibromes sont des tumeurs bénignes issues des filets nerveux. On distingue trois types de neurofibromes :
- Les neurofibromes cutanés : ils ont un aspect de pseudo-hernie, ils sont de la couleur de la peau ou un peu plus rosés, sont dépressibles au toucher, et leur palpation n'entraîne en général aucune douleur. Leur taille est très variable d'un individu à l'autre, et ils n'apparaissent qu'à partir de la puberté. Ils peuvent être enlevés soit par la chirurgie classique soit au laser.
- Les neurofibromes nodulaires : petites tumeurs sous-cutanées qui poussent le long du trajet d'un nerf, en chaînette, ils ont l'aspect de ganglions, et sont douloureux au toucher.
- Les neurofibromes plexiformes : Ce sont des tumeurs souvent de grande taille, qui peuvent être invasifs et donc poser des problèmes de compression de certains organes vitaux. Ils apparaissent très tôt dans la vie, mais leur croissance au moment de la puberté les fait remarquer. Ils sont présents dans la moitié des cas[6]. Ils sont difficilement opérables car très vascularisés, et situés parfois à l'intérieur du corps, d'où l'intérêt d'une IRM, du moins chez l'enfant[7]. Ils peuvent évoluer vers un cancer[8].
Lentigines
Les lentigines sont de petites taches comme des taches de rousseur, mais leur localisation est caractéristique : creux de l'aisselle ou creux de l'aine. Elles sont présentes chez 80 % des personnes atteintes de NF-1.
Nodules de Lisch
Ce sont de petits nodules colorés situés dans l'iris. Difficiles à voir à l'œil nu, on les observe à l'aide d'une lampe à fente. Ils n'entraînent aucun trouble de la vision de l'œil. Ils sont en revanche un bon élément de diagnostic puisque 90 % des adultes atteints de NF-1 en ont. Histologiquement, le nodule de Lisch est un hamartome.
Diagnostic
Des critères de diagnostic ont été établis en 1988. Le diagnostic de neurofibromatose de type I est porté quand au moins deux des signes suivants sont rencontrés[9] :
- six taches ou plus mesurant au minimum 5 mm avant la puberté et 15 mm après la puberté ;
- deux neurofibromes de n'importe quel type ou un fibrome plexiforme ;
- lentigines de la région axillaire ou inguinale ;
- gliome du nerf optique ;
- deux nodules de Lisch ou plus ;
- des anomalies osseuses comme un os sphénoïde dysplasique ou la corticale des os longs très fine ;
- un parent au premier degré atteint par cette maladie.
Gravité
L'importance de cette affection évolutive est extrêmement variable d'un individu à l'autre.
La grande majorité des malades ne présentent que des taches café au lait présentes dès la naissance, puis apparaissant souvent à la puberté, quelques tumeurs bénignes (neurofibromes cutanés) de petite taille et peu nombreuses.
Parfois l'atteinte cutanée est beaucoup plus importante, très nombreux neurofibromes cutanés de taille variable (certains pouvant atteindre 3-4 cm de diamètre) et neurofibromes plexiformes rendant la vie en société difficile à cause du regard des autres si ces tumeurs sont situées sur le visage ou les parties découvertes du corps.
Un certain nombre d'autres problèmes peuvent aussi apparaître :
- déformations de la colonne vertébrale de type scoliose entraînant des douleurs dorsales, parfois la déformation est très importante et nécessite le port d'un corset et même dans les cas les plus graves le recours à la chirurgie orthopédique ;
- déformations corporelles : provoquées par des neurofibromes plexiformes de grande taille ;
- gliome du nerf optique : c'est une tumeur généralement bénigne située au point de rencontre intracrânien des nerfs optiques appelé Chiasma optique ;
- certaines difficultés d'apprentissage : en général les enfants atteints de NF-1 ont une intelligence normale, mais certains rencontrent des difficultés d'apprentissage car ils présentent des problèmes de concentration et une hyper-activité ;
- hypertension artérielle : le risque d'hypertension est augmenté chez les personnes atteintes de NF-1 car ils peuvent présenter un phéochromocytome et des paragangliomes (tumeurs sécrétant des catécholamines) ;
- complications dues à la présence de neurofibromes plexiformes internes volumineux qui peuvent comprimer des organes vitaux ;
- tumeurs cancéreuses, notamment tumeurs malignes des gaines nerveuses.
- pseudarthrose congénitale du tibia (souvent dues à la présence de neurofibromes dans l'os provoquant des fractures) .
Traitement
Aucun traitement certain ne peut actuellement guérir cette maladie, mais la vie des malades peut être améliorée par :
- la destruction des neurofibromes cutanés par électro-coagulation au laser,
- la chirurgie classique, avec :
- l'opération des neurofibromes plexiformes devenus trop gênants ;
- l'opération de déformations trop importantes de la colonne vertébrale.
Le selumetinib permet la régression de certains neurofibromes plexiformes de l'enfant[10].
Divers
Pour toutes les formes de NF, il est vivement conseillé de se faire suivre médicalement la vie entière. Il existe en France une vingtaine d'équipes médicales pluridisciplinaires « neurofibromatoses ».
Les 21 et , l'équipe du professeur Laurent Lantieri de l'hôpital Henri Mondor de Créteil (Val-de-Marne) a effectué une greffe de visage (nez, bouche, menton, joues) sur un malade dont le visage était déformé par des neurofibromes plexiformes volumineux.
Joseph Merrick qui se produisait à Londres sous le surnom de L'Homme éléphant et fut plus tard immortalisé par le film The Elephant Man de David Lynch, a longtemps été considéré comme porteur de la neurofibromatose de type I. Des tests ADN faits dans les années 1990 prouveraient[11],[12],[13], comme les recherches le soupçonnaient depuis longtemps, qu'il était porteur d'une affection très rare, le syndrome de Protée.
Adam Pearson (en), l'acteur qui joue dans le film Under the Skin est atteint de neurofibromatose de type I.
Sources
- (en) J M Friedman, « Neurofibromatosis 1 », in GeneTests: Medical Genetics Information Resource (database online), Copyright, University of Washington, Seattle. 1993-2006[14]
Notes et références
- (en) Hyman SL, Shores A et North KN, « The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. », Neurology, vol. 65, , p. 1037–1044
- (en) Jett K et Friedman JM, « Clinical and genetic aspects of neurofibromatosis 1. », Genet Med, vol. 12, , p. 1–11
- (en) Huson SM, Compston DA, Clark P, Harper PS, « A genetic study of von Recklinghausen neurofibromatosis in south east Wales: I, prevalence, fitness, mutation rate, and effect of parental transmission on severity », J Med Genet. 1989;26:704-711
- (en) Gutmann DH, Wood DL, Collins FS, « Identification of the neurofibromatosis type 1 gene product », Proc Natl Acad Sci U S A. 1991;88:9658-9662
- (en) Altarescu G, Brooks B, Kaplan Y, Eldar-Geva T, Margalioth EJ, Levy-Lahad E, Renbaum P, « Single-sperm analysis for haplotype construction of de-novo paternal mutations: application to PGD for neurofibromatosis type 1 », Hum Reprod, vol. 21, no 8, , p. 2047-51. (PMID 16740526, lire en ligne [html])
- Mautner VF, Asuagbor FA, Dombi E et al. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1, Neuro Oncol, 2008;10:593-598
- Nguyen R, Kluwe L, Fuensterer C, Kentsch M, Friedrich RE, Mautner VF, Plexiform neurofibromas in children with neurofibromatosis type 1: frequency and associated clinical deficits, J Pediatr, 2011;159:652-6555.e2
- Korf BR, Plexiform neurofibromas, Am J Med Genet, 1999;89:31-37
- (en) NIH Consensus Development Conference, « Neurofibromatosis. Conference statement » Arch Neurol. 1988;45:575-8
- Gross AM, Wolters PL, Dombi E et al. Selumetinib in children with inoperable plexiform neurofibromas, N Engl J Med, 2020;382:1430-1442
- (en) MM Cohen Jr. « The Elephant Man did not have neurofibromatosis » Proc Greenwood Genet. Center 1987;6:187-192.
- (en) MM Cohen Jr. « Understanding Proteus syndrome, unmasking the Elephant Man, and stemming elephant fever » Neurofibromatosis 1988;1:260-280.
- (en) MM Cohen Jr. « Further diagnostic thoughts about the Elephant Man » Am J Med Genet. 1988;29:777-782.
- genetests.org
Liens externes
- Page spécifique sur Orphanet
- Moulage ancien de cas extrêmes de Neurofibromatose (collection Péan. Extrait de : Le musée des moulages de l'hôpital Saint-Louis), Idem, autre vue
- Association Neurofibromatoses et Recklinghausen (France)
- Association de la neurofibromatose du Québec
 Portail de la médecine
Portail de la médecine