Règle de Bent
En chimie, la règle de Bent décrit et explique la relation entre l'hybridation d'un atome central dans une molécule et les électronégativités des substituants[1],[2]. La règle est énoncée par Henry Bent en 1961 comme suit : « Le caractère s atomique se concentre aux orbitales dirigées vers les substituants électropositifs. »[2]

La structure chimique d'une molécule est intimement reliée à ses propriétés et sa réactivité. La théorie de la liaison de valence suppose que les structures moléculaires sont le résultat des liaisons covalentes entre les atomes, et que chaque liaison est formée par deux orbitales atomiques en recouvrement qui sont souvent des orbitales hybrides. Dans la version simple de la théorie, les éléments du bloc p sont supposés de ne former que les hybrides spn, où n égale 1, 2, ou 3. En plus, les orbitales hybrides sont supposées toutes équivalentes, c'est-à-dire les n+1 orbitales spn possèdent toutes le même caractère p. Cette approche donne une bonne première description qui peut être améliorée en admettant l'hybridation dite isovalente, où les orbitales hybrides peuvent être fractionnaires et inéquivalentes (spx, x non entiers et inégaux pour deux orbitales différentes). La règle de Bent fournit un estimé qualitatif pour guider la construction de ces orbitales hybrides[3]. Cette règle précise que dans une molécule donnée, un atome central qui est lié à plusieurs groupements d'atomes subira l'hybridation telle que les orbitales avec plus de caractère s se dirigent vers des groupements plus électropositifs, tandis que les orbitales avec davantage de caractère p se dirigent vers des groupements plus électronégatifs. Sans imposer l'hypothèse que toutes les orbitales hybrides sont équivalentes, on peut améliorer la prévision des propriétés telles que la géométrie moléculaire et les énergies des liaisons.
La règle de Bent peut aussi être généralisée aux éléments du bloc d. L'hybridation d'un atome de métal de transition se fait de sorte que les orbitales avec davantage de caractère s se dirigent vers les ligands qui forment des liaisons avec davantage de caractère covalent. De même, les orbitales avec plus de caractère d s'orientent vers les ligands qui forment des liaisons avec davantage de caractère ionique[1].
Historique
Au début des années 1930, peu après le développement initial de la théorie de la liaison de valence, l'application de cette théorie à la structure moléculaire est entamée par Pauling[4], Slater[5], Coulson[6], et autres. En particulier, Pauling introduit la notion de l'hybridation, ou la combinaison des orbitales atomiques pour faire des orbitales hybrides sp, sp2, et sp3. Les orbitales hybrides peuvent fournir des explications des géométries des molécules simples comme le méthane, qui est tétraédrique avec un carbone sp3. Aux années 1940 cependant, on constate des petits écarts par rapport aux géométries idéales[7]. La molécule d'eau est un exemple très bien connu, avec un angle entre les deux liaisons O-H de 104,5°, comparé à la valeur prévue de 109,5°. Pour expliquer ces écarts, on propose que l'hybridation peut former des orbitales inéquivalentes, avec des caractères a et p différents dans une même molécule. En 1947 Arthur Donald Walsh décrit[7] une relation entre l'électronégativité des groupements liés à un atome de carbone et l'hybridation dudit carbone. En 1961 enfin, Henry A. Bent publie une revue importante de la littérature chimique qui relie la structure moléculaire, l'hybridation à l'atome central, et les électronégativités des ligands[2], et la règle de Bent prend son nom de cet ouvrage.
Explication

La règle de Bent est justifiée par le fait que l'orbitale s de valence est plus basse en énergie que les orbitales p de valence du même atome[2]. Les liaisons entre éléments d'électronégativités différentes sont polaires, et la densité électronique d'une liaisons polaire est déplacée vers l'élément le plus électronégatif. À titre d'exemple, considérons les deux types de liaison dans la molécule de fluorométhane (CH3F). Dans les trois liaisons C-H, le carbone est plus électronégatif que l'hydrogène et donc, la densité électronique sera déplacée vers le carbone. L'énergie de ces électrons dépendra beaucoup des orbitales hybrides auxquelles le carbone contribue pour ces liaisons à cause de la densité électronique accrue près du carbone. Si le caractère s de ces orbitales hybrides augmente, l'énergie des électrons liantes est réduite parce que les orbitales s ont une énergie inférieure aux orbitales p.
Par la même logique et sachant que le fluor est plus électronégatif que le carbone, la densité électronique dans la liaison C-F sera déplacée vers le fluor. L'orbitale hybride à laquelle contribue le carbone dans la liaison C-F aura une densité électronique relativement amoindrie par rapport au cas de la liaison C-H. Par conséquent, l'énergie de cette liaison sera moins dépendante de l'hybridation du carbone. En dirigeant une orbitale hybride du caractère p accrue vers le fluor, l'énergie de la liaison C-F n'augmente que peu.
Alors plutôt que diriger des orbitales sp3 équivalentes vers tous les quatre substituants, le carbone déplace de caractère s vers les liaisons C-H, ce qui stabilise ces liaisons beaucoup à cause de la densité électronique accrue proche au carbone, tandis que la diminution de caractère s à la liaison C-F n'augmente son énergie que peu. En accord avec la règle de Bent alors, le caractère s de l'atome central (C) est dirigé vers le ligand électropositif (H) et loin du ligand électronégatif (F).
Le fluorométhane est cas particulier, mais un pareil raisonnement s'applique à toute autre molécule avec un atome central d'un élément représentatif lié à plusieurs atomes d'électronégativités différentes.
Exemples
La règle de Bent aide à expliquer des tendances observées dans la structure moléculaire, les propriétés moléculaires et la reactivité.
Angles de liaison
Les géométries moléculaires dépendent des angles entre les liaisons. Dans la théorie de la liaison de valence, chaque liaison covalente est faite par deux électrons aux orbitales, souvent hybridées, des atomes liés en recouvrement. L'hybridation des orbitales explique pourquoi le méthane est tétraédrique et l'éthylène est plan, par exemple. Cependant, des écarts existent par rapport aux géométries idéales pour l'hybridation spn. Par exemple, les angles de liaison aux molécules d'eau et d'ammoniac sont 104,5° and 107° respectivement, valeurs inférieures à l'angle tétraédrique prévue de 109,5°. En chimie ces différences sont souvent expliquées par la théorie VSEPR, en supposant que les électrons de valence se trouvent en régions localisées et que la répulsion entre les paires d'électrons non liants est supérieure à la répulsion entre les paires liantes.
La règle de Bent fournit une explication alternative des écarts des angles de liaison par rapport aux géométries idéales. La tendance générale de l'hybridation à l'atome centrale en fonction de l'angle de liaison est mise en évidence par les molécules simples de méthane, éthylène, et acétylène. Dans l'ordre, les atomes de carbone de ces molécules dirigent des orbitales sp3, sp2, et sp vers les hydrogènes, et les angles de liaison HCH sont 109,5°, ~120°, and 180°. On s'aperçoit que les angles sont plus petits entre les orbitales atomiques hybridées qui possèdent un caractère p supérieur. Un résultat plus quantitatif est fourni par le théorème de Coulson.

Les exemples au tableau sont présentés à l'article de Bent, qui considère que l'électronégativité du groupement méthyle est inférieure à celle de l'atome d'hydrogène, parce que le remplacement d'un hydrogène par un méthyle réduit la constante de dissociation acide de l'acide formique et aussi de l'acide acétique[2].
| Molécule | Angle de liaison entre substituants |
|---|---|
Diméthyléther | 111° |
Méthanol | 107-109° |
Eau | 104,5° |
Difluorure d'oxygène | 103,8° |
En descendant le tableau, les substituants deviennent plus électronégatifs et l'angle de liaison entre les substituants diminue. Selon la règle de Bent, lorsque les électronégativités des substituants augmentent, les orbitales dirigées vers ces substituants auront un caractère p accrue et les angles de liaison diminuent. Ceci est en accord avec les résultats expérimentaux. Par contre, la théorie VSEPR n'explique pas pourquoi l'angle du diméthyléther est supérieur à 109,5°.
Quant à l'explication de l'angle de liaison de l'eau, un doublet non liant est considéré comme très électropositif, vu qu'il ne possède aucun noyau pour attirer les électrons. Alors la règle de Bent suggère que les doublets non liants occupent des orbitales hybridées avec plus du caractère s. Les orbitales dirigées vers les hydrogènes ont alors plus de caractère p, de sorte que l'angle de liaison entre eux est inférieur à 109.5°. Le même raisonnement s'applique à l'ammoniac.
Longueurs de liaison
Les longueurs de liaison sont aussi reliés à l'hybridation des atomes liés. Le tableau suivant met en évidence que la longueur des liaisons σ carbone-carbone diminue en fonction du caractère s des orbitales au carbones liés[2].
| Molécules | Longueur moyenne de la liaison carbone-carbone |
|---|---|
 | Deux carbones sp3 liésː 1,54 Å |
 | Un carbone sp3 et un carbone sp2 liésː 1,50 Å |
 | Un carbone sp3 et un carbone sp liésː 1,46 Å |
L'addition des substituants électronégatifs permet de changer l'hybridation d'un atome central et alors de varier les longueurs de liaison.
| Molécule | Longueur de la liaison carbone-fluor |
|---|---|
Fluorométhane | 1.388 Å |
 Difluorométhane | 1.358 Å |
 Trifluorométhane | 1.329 Å |
 Tétrafluorométhane | 1.323 Å |
Au fluorométhane le fluor est beaucoup plus électronégatif que l'hydrogène, et alors les orbitales hybridées du carbone qui sont dirigées vers les trois hydrogènes ont plus de caractère s que celles dirigées vers le fluor. Au difluorométhane, il n'y a que deux hydrogènes, et alors il y a moins de caractère s au total dirigé vers eux et il en reste davantage de caractère s dirigé vers les deux fluors. Ceci raccourcit les longueurs de liaison C—F en difluorométhane par rapport au fluorométhane. Cette tendance continue jusqu'au tétrafluorométhane, dont les liaisons C—F possèdent le caractère s au carbone le plus élevé (25%) et les longueurs de liaison les plus courtes dans la série.
La même tendance se trouve pour les analogues chlorés du méthane, quoique l'effet est moins important parce que le chlore est moins électronégatif que le fluor[2].
| Molecule | Average carbon–chlorine bond length |
|---|---|
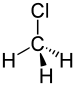 Chlorométhane | 1.783 Å |
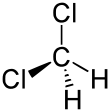 Dichlorométhane | 1.772 Å |
 Trichlorométhane | 1.767 Å |
 Tétrachlorométhane | 1.766 Å |
Ce tableau indique que la grandeur de l'atome du chlore est moins importante que son électronégativité. L'effet stérique seul donnerait la tendance opposée, parce qu'il serait plus favorable que les gros atomes de chlore s'éloignent davantage. Mais cette prévision contredit les résultats expérimentaux, ce qui indique que l'effet de l'hybridation est le principal déterminant de la géométrie, en accord avec la règle de Bent.
Constantes de couplage JCH
La mesure peut-être la plus directe du caractère s d'une orbitale liante entre l'hydrogène et le carbone se sert de la constante de couplage spin-spin 1H−13C. La théorie prévoit que les valeurs de JCH augmentent en fonction du caractère s des liaisons[8],[9].
| Molécule | JCH (des protons méthyliques) |
|---|---|
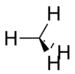 Méthane | 125 Hz |
 Acétaldéhyde | 127 Hz |
 1,1,1-Trichloroéthane | 134 Hz |
 Méthanol | 141 Hz |
 Fluorométhane | 149 Hz |
Lorsque l'électronégativité du substituant augmente, le caractère p dirigé vers le substituant augmente aussi. Ceci laisse davantage de caractère s aux liaisons vers les protons méthyliques, ce qui entraine des constantes de couplage JCH plus élevées.
Effet inductif
L'effet inductif est la transmission de la charge à travers les liaisons covalentes, et la règle de Bent fournit un mécanisme qui l'explique à l'aide des différences d'hybridation[10]. En fonction de l'électronégativité croissante des groupes liés au carbone central au tableau suivant[11], ce carbone devient plus attracteur d'électrons tel qu'indiqué par les constantes polaires des substituants. Ces constantes polaires sont semblables aux valeurs de σ obtenues à partir de l'équation de Hammett; une valeur supérieure correspond à une capacité plus grande de retirer des électrons.
Selon la règle de Bent, si l'électronégativité des groupes substituants augmente, le caractère p dirigé vers ces groupes augmente, ce qui laisse davantage de caractère s à la liaison entre le carbone central et le groupe R. Les orbitales s ayant la densité électronique plus proche au noyau que les orbitales s, la densité électronique à la liaison C−R se déplacera davantage vers le carbone en fonction du caractère s. Le carbone central sera encore plus attracteur d'électrons et retirera davantage des électrons du groupe R[7]. Alors le pouvoir attracteur d'électrons des substituants est transfert au carbone voisin, en accord avec la prévision de l'effet inductif.
| Substituent | Constante polaire de substituant selon Taft (augmente en fonction de l'attraction pour les électrons) |
|---|---|
 Tert-Butyle | -0.30 |
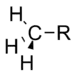 Méthyle | 0.00 |
 Chlorométhyle | 1.05 |
 Dichlorométhyle | 1.94 |
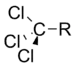 Trichlorométhyle | 2.65 |
Théorie formelle
La règle de Bent rend la théorie de la liaison de valence plus précise. Cette théorie envisage qu'une liaison covalente est constituée de deux électrons situés dans deux orbitales atomiques, normalement hybridées, qui sont en recouvrement sur les atomes liés. L'hypothèse qu'une liaison covalente est combinaison linéaire d'orbitales atomiques sur deux atomes liants seulement est une approximation (voir théorie de l'orbitale moléculaire), mais la théorie de la liaison de valence est assez précis pour avoir une influence majeure (au passé et au présent) sur la compréhension des liaisons chimiques[1].
Dans la théorie de la liaison de valence, chacun de deux atomes contribue une orbitale atomique, et les électrons à la région de recouvrement orbitalaire forment une liaison covalente. Les atomes ne contribuent pas normalement des orbitales hydrogénoïdes pures aux liaisons[4]. Si les atomes ne pouvaient contribuer que les orbitales hydrogénoïdes, alors la structure tétraédrique observée du méthane serait impossible parce que les orbitales 2s et 2p du carbone ne possèdent pas cette géométrie. Cette contradiction parmi d'autres conduit à la proposition de l'hybridation des orbitales. Dans ce cadre, les orbitales atomiques peuvent combiner pour former un nombre pareil de formes et d'énergies différentes. Au cas du méthane, l'orbitale 2s et les trois orbitales 2p du carbone sont hybridées pour faire quatre orbitales sp3 équivalentes, ce qui explique la structure tétraédrique. À l'aide de l'hybridation des orbitales, la théorie de la liaison de valence réussit à expliquer la géométrie et les propriétés d'un très grand nombre de molécules.
Dans la théorie traditionnelle de l'hybridation, les orbitales hybridées sont toutes équivalentes[12]. À savoir les orbitales atomiques s et p sont combinées pour produire quatre orbitales spi3 = 1⁄√4(s+ √3pi), ou trois orbitales spi2 = 1⁄√3(s+ √2pi), ou bien deux orbitales spi = 1⁄√2(s+ pi). Ces combinaisons particulières sont choisies pour satisfaire à deux conditions. D'abord le total des contributions des orbitales s et p doit être pareil avant et après l'hybridation. De plus, les orbitales hybridées doivent être orthogonales les unes aux autres[12]. Si deux orbitales hybridées n'étaient pas orthogonales, elles auraient un recouvrement orbitalaire non nul. Alors si l'une de ces orbitales participait dans une liaison covalente, l'autre aurait aussi une interaction non nulle avec la même liaison, ce qui contredit le principe qu'il n'y a que deux électrons par liaison selon la théorie des liaisons de valence.
Les orbitales hybridées s et p sont construites en supposant d'abord que la première orbitale hybridée est donnée par s+√λipi, où pi est dirigé vers un groupe liant et λi détermine le caractère p de cette orbitale hybridée. Cette formule est une somme pondérée des fonctions d'onde. Ensuite une deuxième orbitale hybridée s+√λjpj est choisie, où pj est dirigée dans une autre direction et λj est le caractère p de la deuxième orbitale. La valeur de λj et la direction de pj sont déterminées pour que l'orbitale résultante soit normalisée et orthogonale à la première orbitale hybridée. L'orthogonalité est requise pour que les deux orbitales hybridées puissent être impliquées en des liaisons covalentes séparées. Le produit scalaire des orbitales orthogonales doit être nul, et alors nous pouvons évaluer le produit scalaire de ces orbitales hybridées.


Ici l'orbitale s est normalisée et alors le produit scalaire . Aussi l'orbitale s est orthogonale aux orbitales pi et pj, de sorte que le deuxième et le troisième terme égalent zéro. Enfin le dernier terme est le produit scalaire de deux orbitales p normalisées à un angle de ωij entre elles, ce qui est égal à cos ωij. Alors l'orthogonalité des orbitales hybridées liantes exige que , ce qui entraîne le théorème de Coulson comme résultat[12] :



Ceci implique que les quatre orbitales atomiques s et p peuvent être hybridées en directions quelconques, pourvu que les coefficients λ de chaque paire des orbitales hybridées satisfont à cette condition pour s'assurer que ces orbitales soient orthogonales.
La règle de Bent prévoit que les orbitales dirigées vers des substituants plus électronégatifs correspondent aux orbitales hybridées s + √λipi avec plus de caractère p, c'est-à-dire aux valeurs supérieurs des coefficients λi. Ainsi si un atome central A est lié à deux groupes X et Y et le groupe Y est plus électronégatif que X, alors A sera hybridé avec λX < λY. Des méthodes théoriques et numériques plus avancées sont requises pour prédire les géométries moléculaires avec précision, mais la règle de Bent fournit un excellent guide qualitative à l'explication des structures moléculaires.
Notes et références
- Weinhold F. et Landis C. L. (2005), Valency and Bonding: A Natural Donor-Acceptor Perspective, Cambridge University Press (ISBN 0-521-83128-8)
- Bent H.A. (1961), An appraisal of valence-bond structures and hybridization in compounds of the first-row elements, Chem. Rev. 61, no 3, p. 275–311, DOI:10.1021/cr60211a005
- Foster J.P. et Weinhold F. (1980), Natural hybrid orbitals, J. Am. Chem. Soc., 102 (24), p. 7211-7218, DOI:10.1021/ja00544a007
- Pauling L. (1931), The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules, J. Am. Chem. Soc., 53 (4), p. 1367–1400, DOI:10.1021/ja01355a027
- Slater J.C. (1931), Directed Valence in Polyatomic Molecules, Phys. Rev., 37 (5), p. 481–489, DOI:10.1103/PhysRev.37.481
- Coulson C. A. (1961), Valence (2e édn.), Clarendon Press (Oxford)
- Walsh A. D. (1947), The properties of bonds involving carbon, Discuss. Faraday Soc., 2, p. 18-25, DOI:10.1039/DF9470200018
- Muller N. et Pritchard D.E.(1959), C13 in Proton Magnetic Resonance Spectra. I. Hydrocarbons, J. Chem. Phys., 31 (3), p. 768–771, DOI:10.1063/1.1730460
- Muller N. et Pritchard D.E.(1959), C13 in Proton Magnetic Resonance Spectra. II. Bonding in Substituted Methanes, J. Chem. Phys., 31 (6), p. 1471–1476, DOI:10.1063/1.1730638
- Bent H.A.(1960), Distribution of atomic s character in molecules and its chemical implications, J. Chem. Educ., 37 (12), p. 616-624, DOI:10.1021/ed037p616
- Taft Jr., R.W.(1957), Concerning the Electron—Withdrawing Power and Electronegativity of Groups, J. Chem. Phys., 26 (1), p. 93-96, DOI:10.1063/1.1743270
- Coulson C. A.(1961), Valence (2e édn.), Clarendon Press (Oxford),p.203-5
 Portail de la chimie
Portail de la chimie