Séquençage shotgun
En génétique, le séquençage shotgun (littéralement séquençage "fusil de chasse") est une méthode utilisée pour séquencer des brins d'ADN aléatoires. On l'appelle ainsi par analogie avec le modèle de tir quasi-aléatoire en pleine expansion d'un fusil de chasse : cette métaphore illustre le caractère aléatoire de la fragmentation initiale de l'ADN génomique où l'on "arrose" tout le génome, un peu comme se dispersent les plombs de ce type d'arme à feu.
La méthode de terminaison de chaîne du séquençage d'ADN ("méthode de Sanger") ne peut être utilisée que pour des brins d'ADN courts de 100 à 1 000 paires de bases. En raison de cette limite de taille, les séquences plus longues sont subdivisées en fragments plus petits qui peuvent être séquencés séparément, et ces séquences sont assemblées pour donner la séquence globale.
Il existe deux méthodes principales pour ce processus de fragmentation et de séquençage. L'arpentage chromosomique (ou "marche des chromosomes") progresse sur le brin entier morceau par morceau, tandis que le séquençage shotgun est un processus plus rapide mais plus complexe qui utilise des fragments aléatoires.
Dans le séquençage shotgun[1],[2], l'ADN est décomposé aléatoirement en de nombreux petits segments, qui sont séquencés en utilisant la méthode de terminaison de chaîne pour obtenir des lectures (reads). Des lectures multiples qui se chevauchent sur l'ADN cible sont obtenues en effectuant plusieurs itérations de cette fragmentation et de ce séquençage. Les programmes informatiques utilisent ensuite les extrémités qui se chevauchent issues de différentes lectures pour les assembler en une séquence continue.
Le séquençage shotgun était l'une des technologies fondatrices à l'origine des projets de séquençage complet du génome.
Exemple
Par exemple, considérons les deux séries de lectures shotgun suivantes :
| Brin | Séquence |
|---|---|
| Original | AGCATGCTGCAGTCATGCTTAGGCTA |
| Première séquence shotgun | AGCATGCTGCAGTCATGCT------- -------------------TAGGCTA |
| Deuxième séquence shotgun | AGCATG-------------------- ------CTGCAGTCATGCTTAGGCTA |
| Reconstruction | AGCATGCTGCAGTCATGCTTAGGCTA |
Dans cet exemple extrêmement simplifié, aucune des lectures ne couvre toute la longueur de la séquence d'origine, mais les quatre lectures peuvent être assemblées dans la séquence d'origine en utilisant le chevauchement de leurs extrémités pour les aligner et les ordonner. En réalité, ce processus utilise d'énormes quantités d'informations qui regorgent d'ambiguïtés et d'erreurs de séquençage. L'assemblage de génomes complexes est en outre compliqué par la grande abondance de séquences répétitives, ce qui signifie que de courtes lectures similaires pourraient provenir de parties complètement différentes de la séquence.
De nombreuses lectures se chevauchant pour chaque segment de l'ADN d'origine sont nécessaires pour surmonter ces difficultés et assembler avec précision la séquence. Par exemple, pour achever le projet du génome humain, la majeure partie du génome humain a été séquencée à une couverture de 12 fois ou plus, c'est-à-dire que chaque base de la séquence finale était présente en moyenne dans 12 lectures différentes. Malgré cela, les méthodes dont on dispose en 2004 n'ont pas réussi à isoler ou à assembler une séquence fiable pour environ 1% du génome (euchromatique) humain[3].
Séquençage shotgun du génome entier
Histoire
L'idée d'utiliser le séquençage shotgun du génome entier pour les petits génomes (4000 à 7000 paires de bases) a été suggérée pour la première fois en 1979[1]. Le premier génome séquencé par le séquençage shotgun était celui du virus de la mosaïque du chou-fleur, publié en 1981[4],[5].
Séquençage à extrémité appariée
Une application plus large a bénéficié du séquençage d'extrémités par paires, connu familièrement sous le nom de séquençage fusil de chasse (shotgun) à double canon. Alors que les projets de séquençage commençaient à prendre en compte des séquences d'ADN plus longues et plus complexes, plusieurs groupes de chercheurs ont commencé à réaliser que des informations utiles pouvaient être obtenues en séquençant les deux extrémités d'un fragment d'ADN. Bien que le séquençage des deux extrémités du même fragment et le suivi des données appariées soient plus lourds que le séquençage d'une seule extrémité de deux fragments distincts, le fait de savoir que les deux séquences étaient orientées dans des directions opposées et avaient environ la longueur d'un fragment indépendamment l'une de l'autre s'est avéré utile pour reconstruire la séquence du fragment cible d'origine.
La première description publiée de l'utilisation du séquençage des extrémités appariées remonte à 1990[6] dans le cadre du séquençage du locus HGPRT humain, bien que l'utilisation des extrémités appariées se soit limitée dans ce cas-ci à combler les lacunes après l'application d'une approche traditionnelle de séquençage shotgun. La première description théorique d'une stratégie de séquençage d'extrémités par paires pures, en supposant des fragments de longueur constante, date de 1991[7]. À l'époque, il y avait un consensus de la communauté que la longueur optimale de fragment pour le séquençage d'extrémité par paire serait trois fois la longueur de lecture de séquence. En 1995, Roach et al.[8] innovent en utilisant des fragments de tailles variables et démontrent qu'une stratégie de séquençage d'extrémités par paires pures serait possible sur des génomes-cibles plus grands. La stratégie a ensuite été adoptée par l'Institut de recherche génomique (TIGR) pour séquencer le génome de la bactérie Haemophilus influenzae en 1995[9], puis par Celera Genomics pour séquencer le génome de Drosophila melanogaster (mouche du vinaigre) en 2000[10] puis le génome humain.
Approche
Pour appliquer cette stratégie, un brin d'ADN de poids moléculaire élevé est cisaillé en fragments aléatoires, sélectionnés en fonction de la taille (généralement 2, 10, 50 et 150 kb) et clonés dans un vecteur approprié. Les clones sont ensuite séquencés à partir des deux extrémités en utilisant la méthode de terminaison de chaîne qui renvoie deux courtes séquences. Chaque séquence est appelée lecture de fin ou lecture 1 et lecture 2, et deux lectures du même clone sont appelées paires d'accouplement. Étant donné que la méthode de terminaison de chaîne ne peut généralement produire que des lectures de 500 à 1 000 bases de long, dans tous les clones sauf les plus petits, les paires d'accouplement se chevauchent rarement.
Assemblage
La séquence d'origine est reconstruite à partir des lectures à l'aide d'un logiciel d'assemblage de séquences. Tout d'abord, les lectures qui se chevauchent sont collectées en séquences composites plus longues appelées contigs. Les contigs peuvent être reliés entre eux en échafaudages en suivant les connexions entre les paires de partenaires . La distance entre les contigs peut être déduite des positions des paires d'accouplement si la longueur moyenne des fragments de la banque est connue et possède une fenêtre de déviation restreinte. Selon la taille de l'espace entre les contigs, différentes techniques peuvent être utilisées pour trouver la séquence dans les espaces. Si l'écart est petit (5-20 kb), alors il faut utiliser une réaction en chaîne par polymérase (PCR) pour amplifier la région, suivie d'un séquençage. Si l'écart est grand (> 20 kb), alors le grand fragment est cloné dans des vecteurs spéciaux tels que des chromosomes artificiels bactériens (BAC), puis on séquence le vecteur.
Avantages et inconvénients
Les partisans de cette approche soutiennent qu'il est possible de séquencer le génome entier en une fois en utilisant de grands tableaux de séquenceurs, ce qui rend l'ensemble du processus beaucoup plus efficace que les approches plus traditionnelles. Les détracteurs soutiennent que bien que la technique puisse séquencer rapidement de grandes régions d'ADN, sa capacité à relier correctement ces régions par la suite est discutable, en particulier pour les génomes avec des régions répétitives. À mesure que les programmes d'assemblage de séquences deviennent plus sophistiqués et que la puissance de calcul devient moins chère, il peut être possible de surmonter cette limitation[réf. nécessaire].
Couverture
La couverture (profondeur ou profondeur de lecture) est le nombre moyen de lectures représentant un nucléotide donné dans la séquence reconstruite. Il peut être calculé à partir de la longueur du génome d'origine (G), du nombre de lectures (N) et de la longueur moyenne de lecture (L) par le biais du calcul . Par exemple, un génome hypothétique avec 2 000 paires de bases reconstruit à partir de 8 lectures avec une longueur moyenne de 500 nucléotides aura une redondance de 2 x (deux fois). Ce paramètre permet également d'estimer d'autres quantités, comme le pourcentage du génome couvert par les lectures (parfois aussi appelé couverture). Une couverture élevée dans le séquençage shotgun est souhaitée car elle peut surmonter les erreurs dans l'appel et l'assemblage des bases. Le domaine de la théorie du séquençage de l'ADN aborde les relations liées à de telles quantités.

Parfois, une distinction est faite entre la couverture de séquence et la couverture physique. La couverture de séquence est le nombre moyen de lectures d'une base (comme décrit ci-dessus). La couverture physique est le nombre moyen de fois qu'une base est lue ou couverte par des lectures de paires d'accouplement[11].
Séquençage shotgun hiérarchique
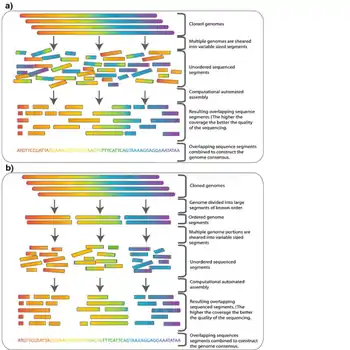
Bien que le séquençage shotgun puisse en théorie être appliqué à des génomes de toutes tailles, son application directe au séquençage de grands génomes (par exemple, le génome humain) a été limitée jusqu'à la fin des années 1990, lorsque les progrès technologiques ont finalement rendu pratique la manipulation de vastes quantités de données complexes générées dans le processus[12]. Historiquement, le séquençage shotgun du génome complet était censé être limité à la fois par la taille des grands génomes et par la complexité ajoutée par le pourcentage élevé d'ADN répétitif (supérieur à 50% pour le génome humain) présent dans les grands génomes[13]. Il n'était pas largement accepté qu'un séquençage shotgun d'un génome complet appliqué à un grand génome fournirait des données fiables. Pour ces raisons, d'autres stratégies qui ont réduit la charge de calcul de l'assemblage de séquences ont dû être utilisées avant le séquençage shotgun. Dans le séquençage hiérarchique, également connu sous le nom de séquençage descendant (top-down), une cartographie génétique à faible résolution du génome est établie avant le séquençage réel. À partir de cette carte, un nombre minimal de fragments qui couvrent l'ensemble du chromosome sont sélectionnés pour le séquençage[14]. De cette façon, la quantité minimale de séquençage et d'assemblage à haut débit est requise.
Le génome amplifié est d'abord cisaillé en morceaux plus gros (50-200 kb) puis cloné dans un hôte bactérien en utilisant des BAC ou des chromosomes artificiels dérivés de P1 (PAC). Comme plusieurs copies du génome ont été cisaillées au hasard, les fragments contenus dans ces clones auront des extrémités différentes, et avec une couverture suffisante (voir la section ci-dessus), il est théoriquement possible de trouver un échafaudage (scaffold) de contigs BAC qui couvre le génome entier. Cet échafaudage est appelé chemin de tuiles (tiling path).

Une fois qu'un chemin de tuiles a été trouvé, les BAC qui forment ce chemin sont cisaillés au hasard en fragments plus petits et peuvent être séquencés en utilisant la méthode shotgun à plus petite échelle.
Bien que les séquences complètes des contigs BAC ne soient pas connues, on connaît en revanche leurs orientations les unes par rapport aux autres. Il existe plusieurs méthodes pour déduire cet ordre et sélectionner les BAC qui composent un chemin de tuiles. La stratégie générale consiste à identifier les positions des clones les uns par rapport aux autres, puis à sélectionner le moins de clones possible requis pour former un échafaudage contigu couvrant toute la zone d'intérêt. L'ordre des clones est déduit en déterminant la manière dont ils se chevauchent[15]. Les clones qui se chevauchent peuvent être identifiés de plusieurs façons. Une petite sonde radioactivement ou chimiquement marquée contenant un site à séquence marquée (STS) peut être hybridée sur une micropuce sur laquelle les clones sont empreints. De cette façon, on peut identifier tous les clones qui contiennent une séquence particulière dans le génome. L'extrémité de l'un de ces clones peut ensuite être séquencée pour donner une nouvelle sonde et on répète le processus dans une méthode appelée arpentage chromosomique.
Alternativement, la banque BAC peut être digérée par des enzymes de restriction. On déduit que deux clones qui ont plusieurs tailles de fragments en commun se chevauchent parce qu'ils contiennent en commun plusieurs sites de restriction espacés de manière similaire[15]. Cette méthode de cartographie génomique est appelée empreinte digitale de restriction car elle identifie un ensemble de sites de restriction contenus dans chaque clone. Une fois qu'on a trouvé le chevauchement entre les clones et qu'on connaît leur ordre par rapport au génome, on séquence par shotgun un échafaudage d'un sous-ensemble minimal de ces contigs qui couvre l'ensemble du génome[14].
Parce qu'il implique d'abord de créer une carte à faible résolution du génome, le séquençage shotgun hiérarchique est plus lent que le séquençage shotgun du génome entier, mais il est cependant moins dépendant des algorithmes informatiques. Néanmoins, le processus de création de banque BAC et de sélection de chemin de tuiles rend le séquençage shotgun hiérarchique lent et laborieux. Maintenant qu'on dispose d'une technologie adaptée et que l'on a démontré la fiabilité des données[13], la vitesse et la rentabilité du séquençage shotgun du génome entier en ont fait la principale méthode de séquençage du génome.
Nouvelles technologies de séquençage
Le séquençage shotgun classique était basé sur la méthode de séquençage de Sanger : c'était la technique la plus avancée pour séquencer les génomes d'environ 1995 à 2005. La stratégie shotgun est toujours appliquée aujourd'hui, mais est basée sur d'autres technologies de séquençage, telles que le séquençage à lecture courte et le séquençage à lecture longue.
Le séquençage à lecture courte ou "nouvelle génération" produit des lectures plus courtes (de 25 à 500 pb) mais plusieurs centaines de milliers ou millions de lectures en un temps relativement court (de l'ordre d'une journée)[16]. Il en résulte une couverture élevée, mais le processus d'assemblage est beaucoup plus intensif en termes de calcul. Ces technologies sont largement supérieures au séquençage Sanger en raison du volume élevé de données et du temps relativement court nécessaire pour séquencer un génome entier[17].
Séquençage shotgun métagénomique
Des lectures de 400 à 500 paires de bases sont suffisantes pour déterminer l'espèce ou la souche à laquelle appartient l'organisme d'où provient l'ADN, à condition que son génome soit déjà connu, en utilisant par exemple un logiciel de classification taxonomique basé sur K-mer. Avec des millions de lectures issues du séquençage de prochaine génération d'un échantillon environnemental, il est possible d'obtenir un aperçu complet de tout microbiome complexe avec des milliers d'espèces, comme la flore intestinale. Les avantages par rapport au séquençage d'amplicon d'ARNr 16S sont :
- le fait qu'il ne se limite pas qu'aux bactéries ;
- la classification au niveau de la souche où le séquençage d'amplicon ne renvoie que le genre ;
- la possibilité d'extraire des gènes entiers et de spécifier leur fonction dans le cadre du métagénome[18].
La sensibilité du séquençage métagénomique en fait un choix attrayant pour une utilisation clinique[19]. Il met cependant l'accent sur le problème de contamination de l'échantillon ou du pipeline de séquençage[20].
Voir aussi
Notes
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Shotgun sequencing » (voir la liste des auteurs).
Références
- Staden, « A strategy of DNA sequencing employing computer programs », Nucleic Acids Research, vol. 6, no 70, , p. 2601–10 (PMID 461197, PMCID 327874, DOI 10.1093/nar/6.7.2601)
- Anderson, « Shotgun DNA sequencing using cloned DNase I-generated fragments », Nucleic Acids Research, vol. 9, no 13, , p. 3015–27 (PMID 6269069, PMCID 327328, DOI 10.1093/nar/9.13.3015)
- Human Genome Sequencing Consortium, « Finishing the euchromatic sequence of the human genome », Nature, vol. 431, no 7011, , p. 931–945 (PMID 15496913, DOI 10.1038/nature03001, Bibcode 2004Natur.431..931H)
- (en) Gardner, Howarth, Hahn et Brown-Luedi, « The complete nucleotide sequence of an infectious clone of cauliflower mosaic virus by M13mp7 shotgun sequencing », Nucleic Acids Research, vol. 9, no 12, , p. 2871–2888 (ISSN 0305-1048, PMID 6269062, PMCID 326899, DOI 10.1093/nar/9.12.2871, lire en ligne)
- (en) Doctrow, « Profile of Joachim Messing », Proceedings of the National Academy of Sciences, vol. 113, no 29, , p. 7935–7937 (ISSN 0027-8424, PMID 27382176, PMCID 4961156, DOI 10.1073/pnas.1608857113)
- Edwards et Caskey, T, « Closure strategies for random DNA sequencing », Methods: A Companion to Methods in Enzymology, vol. 3, no 1, , p. 41–47 (DOI 10.1016/S1046-2023(05)80162-8)
- Edwards, Voss, H., Rice, P. et Civitello, A., « Automated DNA sequencing of the human HPRT locus », Genomics, vol. 6, no 4, , p. 593–608 (PMID 2341149, DOI 10.1016/0888-7543(90)90493-E)
- Roach, Boysen, C, Wang, K et Hood, L, « Pairwise end sequencing: a unified approach to genomic mapping and sequencing », Genomics, vol. 26, no 2, , p. 345–353 (PMID 7601461, DOI 10.1016/0888-7543(95)80219-C)
- Fleischmann, « Whole-genome random sequencing and assembly of Haemophilus influenzae Rd », Science, vol. 269, no 5223, , p. 496–512 (PMID 7542800, DOI 10.1126/science.7542800, Bibcode 1995Sci...269..496F, lire en ligne)
- Adams, « The genome sequence of Drosophila melanogaster », Science, vol. 287, no 5461, , p. 2185–95 (PMID 10731132, DOI 10.1126/science.287.5461.2185, Bibcode 2000Sci...287.2185., lire en ligne)
- Meyerson, Gabriel et Getz, « Advances in understanding cancer genomes through second-generation sequencing », Nature Reviews Genetics, vol. 11, no 10, , p. 685–696 (PMID 20847746, DOI 10.1038/nrg2841)
- Dunham, I. Genome Sequencing. Encyclopedia of Life Sciences, 2005. DOI:10.1038/npg.els.0005378
- Venter, J. C. ‘’Shotgunning the Human Genome: A Personal View.’’ Encyclopedia of Life Sciences, 2006.
- Gibson, G. and Muse, S. V. A Primer of Genome Science. 3rd ed. P.84
- Dear, P. H. Genome Mapping. Encyclopedia of Life Sciences, 2005. DOI:10.1038/npg.els.0005353.
- Karl, « Next Generation Sequencing: From Basic Research to Diagnostics », Clinical Chemistry, vol. 55, no 4, , p. 41–47 (PMID 19246620, DOI 10.1373/clinchem.2008.112789)
- Metzker, « Sequencing technologies - the next generation », Nat Rev Genet, vol. 11, no 1, , p. 31–46 (PMID 19997069, DOI 10.1038/nrg2626, lire en ligne)
- Roumpeka, « A review of bioinformatics tools for bio-prospecting from metagenomic sequence data », Frontiers in Genetics, vol. 8, , p. 23 (PMID 28321234, PMCID 5337752, DOI 10.3389/fgene.2017.00023)
- Gu, « Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection », Annual Review of Pathology: Mechanisms of Disease, vol. 14, , p. 319–338 (PMID 30355154, PMCID 6345613, DOI 10.1146/annurev-pathmechdis-012418-012751)
- Thoendel, « Impact of contaminating DNA in whole genome amplification kits used for metagenomic shotgun sequencing for infection diagnosis », Journal of Clinical Microbiology, vol. 55, no 6, , p. 1789–1801 (PMID 28356418, PMCID 5442535, DOI 10.1128/JCM.02402-16)
Lectures complémentaires
- (en) "Le séquençage shotgun arrive à maturité"par Tabitha Powledge, 2002, sur TheScientist
- (en) "Le séquençage shotgun permet de trouver des nano-organismes : une sonde de drainage minier acide révèle un archée de la taille d'un virus qu'on n'attendait pas" Communiqué de presse de l'Université de Californie à Berkeley, 2006
Liens externes
- Cet article contient du matériel du domaine public issu du document du NCBI appelé "NCBI Handbook".
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire