SN1
La substitution nucléophile monomoléculaire, plus couramment appelée SN1 est un mécanisme réactionnel en chimie organique. C'est un mécanisme limite, au sens où des réactions chimiques « naturelles » usant de ce type de mécanisme ne se font jamais entièrement selon ce mécanisme, mais à un certain pourcentage. Le mécanisme limite « opposé » est la SN2. Ces deux mécanismes sont utilisés pour décrire la réaction :
- R-GP + Nu− = R-Nu +GP−
où GP est le groupe partant (aussi appelé nucléofuge), Nu est le nucléophile, et R un radical alkyle ou aryle.
Cinétique
La cinétique de la réaction est de type v = k[R-GP]. Ce type de réaction est dit monomoléculaire, car l'étape cinétiquement limitante dans le mécanisme réactionnel ne fait intervenir qu'une seule molécule du côté des réactifs, donc que la cinétique de la réaction, dans l'approximation de l'étape cinétiquement déterminante (limitante), ne dépend que de la concentration de cette espèce.
- [R-GP] représente la concentration en substrat en mol.L−1
- v représente la vitesse de la réaction de substitution nucléophile. Elle s'exprime en mol.L−1.s−1
- k est la constante de vitesse; son unité se déduit de celle des 2 autres termes: elle s'exprime donc en s−1
Mécanisme
La réaction se fait en deux étapes.
1re étape
Le groupe partant GP réagit sur lui-même. La liaison R-GP se rompt pour donner un carbocation R+ et un ion GP−. C'est une réaction endothermique, très lente et déterminante pour la cinétique de la réaction.
Exemple avec du 3-chloro-3-éthylpentane : ici c'est le groupe Cl qui joue le rôle de groupe partant. Les lobes en bleu représentent l'orbitale p vacante.
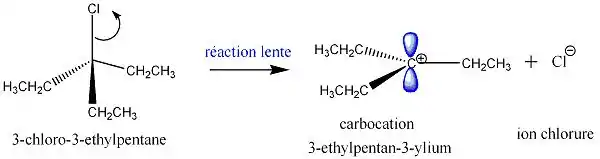
2e étape
Le nucléophile Nu− réagit immédiatement (étape rapide) avec le carbocation juste après sa formation. Le nucléophile peut attaquer le carbocation, qui est de géométrie trigonale plane, en dessous ou au-dessus du plan.
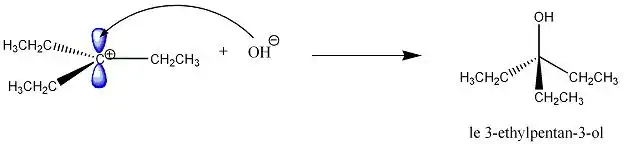
Avec l'exemple précédent, le produit est le même quel que soit le côté attaqué car le 3-chloro-3-ethylpentane est une molécule achirale.
Stéréochimie
Lors d'une SN1, dans le cas où le réactif principal est une molécule chirale, il y a formation d'un mélange racémique (50 % de produit de configuration S + 50 % de produit de configuration R), ou bien d'un mélange de diastéréoisomère optiquement actif si la molécule comporte plusieurs carbones asymétriques.
La première étape est identique que la molécule soit chirale ou non. C'est lors de la seconde étape que la configuration du réactif entre en jeu. En effet, avec un réactif chiral, le nucléophile peut attaquer le carbocation formé à deux endroits différents. Statistiquement il y a autant de chance que le nucléophile se place sur un côté plutôt qu'un autre d'où la formation d'autant de composé R que de composé S.
Exemple avec le (S)-3-chloro-méthylhexane :
1re étape
Le chlore réagit sur lui-même pour former l'ion chlorure et un carbocation (étape cinétiquement limitante).
2e étape
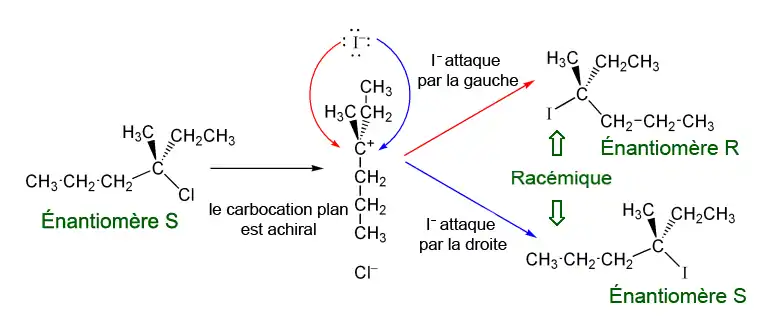
- Première possibilité :
Le nucléophile ici joué par I− attaque le carbocation, qui forme un plan trigonal, par la gauche. Le composé formé est de configuration R.
- Deuxième possibilité :
Le nucléophile I− attaque le carbocation par la droite. Le composé formé est de configuration S.
On constate une racemisation des produits.
Influence de quelques facteurs
Classe de R
Expérimentalement, on observe, lors d'un mécanisme SN1, que la vitesse augmente quand on passe des composés primaires, aux secondaires, puis aux tertiaires. L'influence de R est donc totalement opposée à celle dans le mécanisme de SN2. On peut ainsi schématiser la réactivité:
Cet ordre peut être interprété par une stabilité croissante de l’intermédiaire réactionnel (carbocation dans le cas d'une SN à partir d'un halogénoalcane), donc de la diminution de l'énergie d'activation , ainsi, une augmentation de la constante de vitesse de l'étape de formation de cet intermédiaire réactionnel qui détermine la vitesse de la réaction. (On peut schématiser ceci par le fait que plus l’intermédiaire réactionnel est stable, plus il est "facile" de le former, donc plus la réaction est rapide, cf le postulat de Hammond)
Réactif nucléophile
La « force » du nucléophile, sa concentration ou sa taille, n'ont pas d'influence sur la vitesse de réaction, puisqu'il n'intervient pas dans l'étape cinétiquement limitante. Il n'a donc, en théorie, aucune influence sur la vitesse de la réaction.
Polarité du solvant
L'intermédiaire réactionnel est un ion (carbocation dans le cas de l'halogénoalcane en tant que réactif). Il est donc stabilisé par un solvant polaire, alors que le réactif initial ne l'est quasiment pas, de par sa globale neutralité électrique. Donc la vitesse de la réaction augmente avec la polarité du solvant pour un mécanisme de type SN1.
Nature du nucléofuge (groupe partant, GP)
La liaison R-GP se rompt d'autant plus facilement qu'elle est polarisable. Pour cela il faut qu'elle soit la plus longue possible. Dans le cadre d'une SN1 sur un halogénoalcane, la vitesse augmente donc de R-F à R-I.
Parmi les halogènes, on peut finalement classer la nucléofugacité comme ceci :

Ainsi, l'iode est un meilleur nucléofuge que le chlore grâce à sa plus grande polarisabilité. Par conséquent l'ordre de nucléophilie est inversement proportionnel à l'électronégativité.
Concurrence
Cette réaction est en concurrence avec l'autre substitution nucléophile (SN2), mais aussi avec les réactions d'éliminations, en particulier la réaction d'élimination monomoléculaire (E1), dont les conditions de réalisation sont proches.
 Portail de la chimie
Portail de la chimie