Syndrome de Prader-Willi
Le syndrome de Prader-Willi (SPW) est une maladie génétique rare qui entraîne un grand nombre de symptômes, de nature et de degrés très variables suivant les individus. Ce syndrome est notamment caractérisé à la naissance par une hypotonie sévère, un trouble de la croissance, suivis par l'apparition d'une hyperphagie (problème avec l'hormone de satiété, l'enfant ne sent pas qu'il a assez mangé) pendant l'enfance (entre 2 ans et 8 ans en général), qui, sans prise en charge adaptée, peut conduire au développement d’une obésité morbide.
Pour les articles homonymes, voir Willi.
| Médicament | Somatrem (en) |
|---|---|
| Spécialité | Génétique médicale, pédiatrie et neurologie |
| CIM-10 | Q87.1 |
|---|---|
| CIM-9 | 759.81 |
| OMIM | 176270 |
| DiseasesDB | 10481 |
| MedlinePlus | 001605 |
| eMedicine | 947954 |
| MeSH | D011218 |
| GeneReviews | |
| Patient UK | Prader-willi-syndrome-pro |
![]() Mise en garde médicale
Mise en garde médicale
Le syndrome de Prader-Willi est classé parmi les maladies rares affectant un cas sur 25 000 naissances environ.
Historique
Le syndrome est décrit en 1956 par trois médecins suisses Andrea Prader (en), Alexis Labhart (es) et Heinrich Willi (en) de l'Hôpital pédiatrique de Zurich (de), sous la direction de Guido Fanconi (1892-1979), d'où l'appellation initiale de syndrome de Prader-Labhart-Willi[1].
%252C_de_Juan_Carre%C3%B1o_de_Miranda..jpg.webp)
Ces auteurs établissent le syndrome à partir de l'observation de 9 enfants réunissant quatre caractéristiques communes[1] :
- Petite taille ;
- Retard mental ;
- Obésité ;
- Petitesse des mains et des pieds.
En 1961, Prader et Willi révisent cette définition en signalant d'autres caractéristiques et en insistant sur l'hypotonie du début de la vie et la survenue plus tardive d'un diabète[1].
L'appellation moderne de syndrome de Prader-Willi (disparition de Labhart ou Labhardt) pourrait aussi s'expliquer par le fait qu'aux États-Unis, les dénominations à trois patronymes sont jugées trop longues[2].
En 1967, une observation du XIXe siècle, publiée par le médecin britannique John Langdon-Down (1826-1896) à propos d'un cas de « polysarcie » (obèse naine âgée de 14 ans avec retard mental) a été jugée rétrospectivement comme le premier cas médicalement décrit de syndrome de Prader-Willi[1],[3].
Un autre diagnostic rétrospectif historique est fait en 2000 à propos du tableau La Monstrua denuda de Juan Carreno de Miranda (1614-1685) qui serait « presque certainement » un cas de syndrome de Prader-Willi[4].
En 1981, la principale base génétique de la maladie est précisée : une perte (délétion) de gènes sur le bras long du chromosome 15[5].
Dans les années 1990-2000, avec les progrès de génétique moléculaire, le diagnostic peut être fait de façon plus précoce, dès les premiers mois de vie, voire en diagnostic prénatal. De même la prise en charge sociale et sanitaire des patients s'améliore avec l'existence d'associations de patients en France et dans le monde[6].
On trouve parfois les appellations Prader Willi ou Willi Prader (sans le tiret) par ceux qui prennent Willi pour le prénom de Prader[2].
Épidémiologie
Le SPW est une maladie rare : 1 sur 20 000 à 25 000 naissances environ (extrêmes : 1 sur 8 000 à 1 sur 30 000 selon les études). En 2016, le nombre de cas mondiaux est estimé à 400 000, dont 20 000 aux États-Unis[7].
En Europe, l'incidence annuelle est estimée à 1 pour 30 000 naissances, et la prévalence est estimée à 1 sur 50 000 habitants[8].
La plupart des cas sont sporadiques, touchant à peu près également garçons et filles. Les cas familiaux, avec un risque de transmission pouvant aller jusqu'à 50 %, sont très rares[5],[8].
Il n'y a pas de réelles différences ethniques, mais aux États-Unis, les noirs ont été moins signalés que les blancs[7].
Le SPW est la maladie la plus fréquente (« chef de file ») des obésités morbides d'origine génétique[7].
Étiologie
Dans 98 % des cas, l'anomalie génétique s'est produite lors du développement de l'enfant : elle n'a pas été transmise par les parents[8].

La cause de cette maladie est génétique. Les gènes existent en double exemplaire par paire de chromosomes : une copie d'origine maternelle et une copie d'origine paternelle. La plupart des gènes sont actifs ou inactifs de la même façon pour leurs deux copies, mais certains gènes, notamment ceux du développement, nécessitent que l'une des copies parentales soit réprimée, l'autre pouvant être exprimée. Cette répression génique s'appelle l'empreinte parentale[8].
Dans le cas du syndrome de Prader-Willi (SPW), les gènes en cause sont situées sur le bras long du chromosome 15 (région 15q11-q13). Normalement, seule la copie paternelle de cette région est active, la copie maternelle restant non fonctionnelle ou silencieuse. Or dans le SPW, la copie paternelle est absente ou inactive tandis que la copie maternelle reste silencieuse[8].
Les principaux mécanismes responsables de cette inactivation sont[8] :
- dans 60 à 70 % des cas, perte (délétion) du locus q11.2-q13 du chromosome 15 paternel ;
- dans 25 à 30 % des cas, disomie maternelle de la paire de chromosome 15 (deux copies d'origine maternelle). Ce mécanisme tend à devenir plus fréquent, car il est en rapport avec l'âge maternel[6].
- dans moins de 5 % des cas, il s’agit d’un défaut d’expression de la région SPW bien que présente (anomalie d'empreinte).
Description clinique
Les manifestations sont caractéristiques, mais la sévérité et le moment d'apparition des troubles sont variables d'un sujet à l'autre.
Période prénatale
Le fœtus bouge moins que la normale, avec parfois un liquide amniotique en excès (hydramnios)[8].
Hypotonie
L’hypotonie à la naissance est un signe constant. Cette hypotonie s’accompagne d’une faiblesse du réflexe de succion avec des difficultés à s'alimenter, une perte de masse et de force musculaire, et un retard staturo-ponderal. L'enfant pleure peu et sans cris[6].
Cette hypotonie avec trouble de la déglutition expliquent en partie la fréquence des infections respiratoires (rhume, otite, bronchite...). Cette hypotonie s'améliore le plus souvent dans les premières années de la vie, mais une fatigabilité à l'effort peut persister[8].
Syndrome dysmorphique
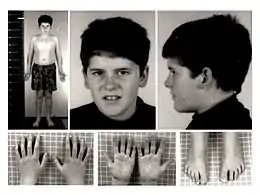
L'enfant présente parfois des caractéristiques faciales particulières, comme le front étroit, les yeux en amande, un strabisme. La bouche est petite avec les coins des lèvres tournés vers le bas, la lèvre supérieure fine, la salive épaisse et peu abondante. D'autres aspects peuvent se révéler progressivement par la suite comme une petite taille, la petitesse des mains et des pieds, l'hypoplasie de l'émail dentaire qui favorise les caries[8],[9].
Hypogonadisme
L’hypogonadisme est présent chez tous les patients. Chez les garçons, il est évident par absence de descente des testicules ou cryptorchidie (bourses petites, vides et peu pigmentées) et pénis anormalement petit (micropénis). Chez les filles, le clitoris et les petites lèvres sont peu développées[8].
Troubles alimentaires
Ils apparaissent progressivement vers un ou deux ans, mais à un rythme différent selon les enfants. Le poids augmente d'abord sans que l'appétit augmente, puis l'hyperphagie apparait vers l'âge de 8 ans par manque de sensation de satiété. Elle peut aboutir à une obésité qui touche surtout l'abdomen, les fesses et les cuisses, avec des risques de complications graves (diabète, problèmes cardiovasculaires et respiratoires...)[8],[6].
Ces troubles se rapprochent d'une addiction ou d'une tendance compulsive à consommer de la nourriture en quantité très importante[8].
Troubles endocriniens
Il existe des anomalies hypothalamiques et du fonctionnement des structures du cerveau qui régulent l'appétit et le comportement alimentaire. Ces anomalies s'accompagnent d'un déficit en hormone de croissance et en hormones sexuelles[6].
En l'absence de traitement, ces troubles se manifestent par une taille plus petite que la moyenne (1,55 m pour les hommes et 1,48 m pour les femmes), et par des troubles de la puberté (hypogonadisme, voix fluette chez les garçons, seins peu développées et règles faibles et irrégulières chez les filles). Dans un petit nombre de cas, la puberté peut être précoce, mais en restant incomplète[8].
Dans les deux sexes, l'infertilité est fréquente.
Troubles cognitifs et du comportement
Lorsqu'il existe, le déficit intellectuel reste modéré et variable d'un enfant à l'autre. La plupart des enfants, même sans retard mental, ont des difficultés d'apprentissage : acquisition retardée du langage, difficultés à bien articuler et comprendre les mots (notamment le sens figuré ), à communiquer (troubles de la pragmatique )[6].
Les troubles du comportement apparaissent vers l'âge de 3 ou 4 ans : rigidité mentale avec obstination, crises brutales de colère incontrôlable (peur du changement, fatigue ou frustration, rapport avec la nourriture)[8].
L'enfant s'attache à des rituels ou des idées fixes, des manies répétitives et des troubles obsessionnels compulsifs. Il peut avoir un comportement autoagressif à type d'automutilation (grattage de la peau ou des muqueuses), ainsi que des troubles du sommeil (apnées du sommeil, endormissement involontaire dans la journée)[8].
Des troubles du spectre autistique (TSA) sont présents (chez 25 à 41 % des cas de SPW), mais ces diagnostics manquent de précision (car basés sur des tests questionnaires adressés aux parents), ce qui peut nuire aux interventions et recherches futures. Ces enfants devraient être évalués par observation directe [10].
Diagnostic
Diagnostic prénatal
Les tests les plus récents de dépistage prénatal non invasif (DPNI) réalisés par un simple prélèvement de sang maternel pour les femmes enceintes visent à isoler l'ADN fœtal et à identifier des anomalies chromosomique et notamment les risques de syndrome de Prader-Willi.[11]
Diagnostic positif

Clinique
Le diagnostic clinique peut être suspecté selon des critères définis en 1993 (majeurs valant 1 point et mineurs valant un demi-point). Ces critères cliniques sont variables au cours de la vie : avant 3 ans, cinq points sont nécessaires, dont quatre critères majeurs. Après 3 ans, huit points sont nécessaires, dont cinq critères majeurs[9].
Ces critères cliniques ont été révisés en 2001, et affinés par la suite en fonction de l'âge de diagnostic (0 à 2 ans, de 2 à 6 ans, de 6 à 12 ans, de 13 ans à l'âge adulte). Ils ont été développés à une époque où les moyens de génétique moléculaire n'étaient pas encore largement disponibles. Ils restent utiles pour le clinicien pour orienter le patient vers une confirmation biologique (éviter les faux diagnostics par excès ou par défaut)[12],[13].
Biologique
Des examens de laboratoire spécialisé sont indispensables pour confirmer le diagnostic. Dans le premier temps, une analyse génétique est réalisée (étude de la méthylation de la région PWS/AS du chromosome 15). Dans la presque totalité des cas, les patients atteints de Prader-Willi ont des anomalies isolées de la méthylation. Un profil normal de méthylation de cette région exclut à 99 % le diagnostic de syndrome de Prader-Willi[8],[6].
Une fois le diagnostic confirmé, d'autres méthodes sont utilisées pour préciser le mécanisme génétique exact et permettre un conseil génétique ou une prise en charge[6].
Diagnostic différentiel
De nombreux troubles peuvent ressembler en partie à des manifestations apparentes (phénotype) de Prader-Willi[13].
Le craniopharyngiome est une tumeur embryonnaire qui peut léser l'hypothalamus et simuler des manifestations de SPW. D'autre part, un environnement traumatisant (stress psycho-social) du développement de l'enfant peut aussi conduire à une petite taille avec comportement hyperphagique[13].
Hypotonie
L’hypotonie à la naissance est aussi présente dans :
- Anomalie chromosomique par délétion : du Xq27.2-ter, du 6q16.2, du 1p36, du 10q26 ;
- Syndrome de l'X fragile ;
- Syndrome de Rett ;
- Syndrome d'Angelman.
L'hypotonie du nourrisson peut être due à une infection néonatale, des myopathies et neuropathies[13].
Obésité
L'obésité du SPW est une obésité syndromique (par opposition à l'obésité isolée) car elle s'inscrit dans un ensemble d'autres manifestations.
L’association retard de développement et obésité, avec ou sans hypogonadisme, se rencontre dans[13] :
Complications et pronostic
Les principales complications sont en rapport avec l'hyperphagie et l'obésité. Si l'accès à la nourriture et la prise de poids ne sont pas suffisamment contrôlés, il y a risque d'apparition de : diabète sucré, ostéoporose, stéatose hépatique, difficultés respiratoires (hypoventilation, apnée du sommeil...), insuffisance cardiaque droite, cellulite et varices (ulcère veineux)[7].
Ces troubles peuvent s'accompagner de troubles de la mastication et de la déglutition, de troubles moteurs du tube digestif. Les patients présentent un seuil élevé à la douleur digestive et au réflexe de vomissement. Ils ne ressentent guère la distension abdominale et les souffrances d'organe. Cette situation expose à un risque élevé de fausse route et de suffocation[7], de rupture gastrique[6].
Aux États-Unis, une étude publiée en 2007 a montré que près 8 % des décès de patients atteints de SPW sont dus à des suffocations alimentaires surtout par hot dog[13].
Une intoxication par l'eau (soif non contrôlée) est possible avec déséquilibre électrolytique dont l'hyponatrémie de dilution[7].
D'autres complications sont[8],[13] :
- les déviations de la colonne vertébrale (scoliose, cyphose), articulation de la hanche (luxation de hanche) ;
- Problèmes visuels : strabisme, myopie... ;
- Troubles de régulation de la température : fièvre inexpliquée ou absence de fièvre en cas d'infection.
Le pronostic dépend de la prise en charge précoce et du traitement. Le taux de mortalité est estimé à 3 % par an, mais peut être réduit de moitié par amélioration du suivi. Dans l'enfance, les causes de décès de SPW sont principalement respiratoires et infectieuses, et à l'âge adulte ce sont les problèmes cardiovasculaires liés à l'obésité[13].
Avec un diagnostic et un traitement précoces, les patients peuvent avoir une espérance de vie moyenne dépassant les 60 ans lorsque les comorbidités et le poids sont bien contrôlés[14].
Traitement
Il n'existe aucun traitement spécifique au syndrome de Prader-Willi. Seule la prévention des symptômes (traitements hormonaux, régime équilibré, orthophonie, etc.) permet d'améliorer la qualité de vie des patients. La prise en charge se fait par équipe pluridisciplinaire de soignants selon les troubles présentés.
Chez le nourrisson
L'hypotonie et les troubles de la succion-déglutition à la naissance, s'ils sont sévères, peuvent rendre l'alimentation impossible. L'enfant est hospitalisé et nourri à l'hôpital par sonde nasogastrique, voire par gastrostomie, durant les premières semaines de vie.
Le traitement par hormone de croissance recombinante peut débuter dès la première année de vie. C'est le traitement standard de base du SPW qui améliore le phénotype des enfants et adultes atteints de SPW, avec ou sans déficit d'hormone de croissance[7].
Les anomalies génitales sont traitées par chirurgie.
De l'enfance à l'âge adulte
Le traitement par hormone de croissance est poursuivi jusqu'à la fin de la croissance, parfois au delà dès lors que le déficit hormonal persiste.
Un traitement par hormones sexuelles est démarré en général à l'âge de la puberté. Ce traitement a été discuté au début des années 1990, à cause d'éventuels effets secondaires, mais depuis il est jugé efficace et généralement bénéfique en l'absence de contre-indications[2],[5].
Le contrôle de l'alimentation et du poids est indispensable et doit commencer avant l'apparition de l'obésité. L'éducation alimentaire précoce et le contrôle strict de l'accès à la nourriture sont essentiels.
Des médicaments psychotropes peuvent être prescrits à faible dose pour les troubles du comportement et les manifestations psychiatriques.
Pour limiter et prévenir le handicap, il est fait appel aux professionnels : kinésithérapie, orthophonie, diététique, orthopédie, soutien psychologique de l'enfant et de sa famille[8].
Associations de familles
Dans le monde
Des associations de patients, présentes dans 102 pays en 2015, sont regroupées en une organisation internationale, l'IPWSO International Prader-Willi Syndrome Organisation qui tient des congrès internationaux depuis 1991, où se réunissent familles, soignants et chercheurs[6].
Depuis 2003, il existe une fondation dédiée pour la recherche, la FPWR Foundation for Prader-Willi Research[6].
France
L'association Prader-Willi France, qui s'adresse notamment aux parents d'enfants porteurs du SPW, est en lien avec la recherche médicale et les professionnels de santé spécialisés. Elle propose en particulier un guide[15] ainsi qu'une écoute par le biais de correspondants régionaux[16], et permet aux membres de l'association d'être tenus informés des dernières avancées médicales concernant le SPW lors de la journée nationale annuelle[17].
Notes et références
- « Prader-Labhardt-Willi syndrome », sur www.whonamedit.com (consulté le )
- Cees Noordam, Charlotte Höybye et Urs Eiholzer, « Prader–Willi Syndrome and Hypogonadism: A Review Article », International Journal of Molecular Sciences, vol. 22, no 5, (ISSN 1422-0067, PMID 33800122, PMCID 7962179, DOI 10.3390/ijms22052705, lire en ligne, consulté le )
- (en) O. Conor Ward, « Prader-Willi syndrome », The Lancet, vol. 356, no 9244, , p. 1856 (ISSN 0140-6736 et 1474-547X, PMID 11117944, DOI 10.1016/S0140-6736(05)73324-9, lire en ligne, consulté le )
- Mary Jones, « Newsletter 2000 (A case study) », sur web.archive.org, (consulté le ), p. 8
- Luigi Napolitano, Biagio Barone, Simone Morra et Giuseppe Celentano, « Hypogonadism in Patients with Prader Willi Syndrome: A Narrative Review », International Journal of Molecular Sciences, vol. 22, no 4, (ISSN 1422-0067, PMID 33671467, PMCID 7922674, DOI 10.3390/ijms22041993, lire en ligne, consulté le )
- Maïthé Tauber, Denise Thuilleaux et Éric Bieth, « Le syndrome de Prader-Willi en 2015 », Medecine Sciences: M/S, vol. 31, no 10, , p. 853–860 (ISSN 1958-5381, PMID 26481024, DOI 10.1051/medsci/20153110011, lire en ligne, consulté le )
- Merlin G. Butler, Jennifer L. Miller et Janice L. Forster, « Prader-Willi Syndrome - Clinical Genetics, Diagnosis and Treatment Approaches: An Update », Current Pediatric Reviews, vol. 15, no 4, , p. 207–244 (ISSN 1573-3963, PMID 31333129, PMCID 7040524, DOI 10.2174/1573396315666190716120925, lire en ligne, consulté le )
- « Le syndrome de Prader-Willi » [PDF], sur Orpha.net (consulté le ).
- (en) Vanja A. Holm, Suzanne B. Cassidy, Merlin G. Butler et Jeanne M. Hanchett, « Prader-Willi Syndrome: Consensus Diagnostic Criteria », Pediatrics, vol. 91, no 2, 1993-02-xx, p. 398–402 (ISSN 0031-4005, PMID 8424017, PMCID 6714046, lire en ligne, consulté le )
- (en) Elisabeth M. Dykens, Elizabeth Roof, Hailee Hunt-Hawkins et Nathan Dankner, « Diagnoses and characteristics of autism spectrum disorders in children with Prader-Willi syndrome », Journal of Neurodevelopmental Disorders, vol. 9, no 1, , p. 18 (ISSN 1866-1947 et 1866-1955, PMID 28592997, PMCID PMC5458479, DOI 10.1186/s11689-017-9200-2, lire en ligne, consulté le )
- (en) « Nifty Test »
- (en) Meral Gunay-Aygun, Stuart Schwartz, Shauna Heeger et Mary Ann O'Riordan, « The Changing Purpose of Prader-Willi Syndrome Clinical Diagnostic Criteria and Proposed Revised Criteria », Pediatrics, vol. 108, no 5, , e92–e92 (ISSN 0031-4005 et 1098-4275, PMID 11694676, DOI 10.1542/peds.108.5.e92, lire en ligne, consulté le )
- Daniel J. Driscoll, Jennifer L. Miller, Stuart Schwartz et Suzanne B. Cassidy, « Prader-Willi Syndrome », dans GeneReviews®, University of Washington, Seattle, 1993 (revised 2017) (PMID 20301505, lire en ligne)
- Maria A. Fermin Gutierrez et Magda D. Mendez, « Prader-Willi Syndrome », dans StatPearls, StatPearls Publishing, (PMID 31985954, lire en ligne)
- « Guide de pratiques partagées », sur guide-prader-willi.fr (consulté le )
- « Première visite - Prader-Willi France », sur www.prader-willi.fr (consulté le )
- « Vie Nationale - Prader-Willi France », sur www.prader-willi.fr (consulté le )
Bibliographie
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 176270
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 182279
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 602117
- (en) Suzanne B Cassidy, Stuart Schwartz, Prader-Willi Syndrome in GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005
Liens externes
- (en-GB) « International Prader-Willi Syndrome Organisation », sur IPWSO (consulté le )
- (en) FPWR, « Foundation for Prader-Willi Research », sur www.fpwr.org (consulté le )
- Association Prader-Willi France
- « Prader-Willi Belgique », sur www.praderwilli-belgium.be (consulté le )
- (de-CH) « Prader-Willi Suisse », sur Prader-Willi-Syndrom Vereinigung Schweiz (consulté le )
- (fr/en) Fondation canadienne pour la recherche sur le syndrome de Prader-Willi
 Portail du handicap
Portail du handicap  Portail de la médecine
Portail de la médecine