Syndrome de Rothmund-Thomson
Le syndrome de Rothmund-Thomson (SRT) est une maladie génétique rare caractérisée par une transmission autosomique récessive.
| Spécialité | Génétique médicale |
|---|
| CIM-9 | 757.33 |
|---|---|
| OMIM | 268400 |
| DiseasesDB | 29891 |
| eMedicine | 1112093 |
| MeSH | D011038 |
![]() Mise en garde médicale
Mise en garde médicale
| Syndrome de Rothmund-Thomson | |
| Référence MIM | 268400 |
|---|---|
| Transmission | Récessive |
| Chromosome | 8q24.3 |
| Gène | RECQL4 |
| Empreinte parentale | Non |
| Mutation | Ponctuelle |
| Nombre d'allèles pathologiques | 28 |
| Nombre de cas | Plus de 300 |
| Maladie génétiquement liée | Syndrome Rapadilano |
| Diagnostic prénatal | Possible |
| Liste des maladies génétiques à gène identifié | |
Cause
L'un des gènes en cause est le RECQL4 codant une hélicase à acide désoxyribonucléique[1]. Une mutation du gène donnant une protéine non fonctionnelle serait responsable des 2/3 des patients atteints et serait corrélée avec une augmentation du risque de formation d'un ostéosarcome[2].
Historique
Le SRT fut décrit pour la première fois en 1868 par un ophtalmologiste allemand, Auguste Rothmund, qui trouva dans les membres d’une même famille de l’érythème et des cataractes juvéniles. En 1923, un dermatologue anglais, Sydney Thomson, appela « Poïkiloderma congenitale » l’érythème si caractéristique du SRT. Finalement, c’est en 1957 que William Taylor suggéra que les deux maladies faisaient partie d'une même maladie et décida d’unir les deux noms pour en faire le syndrome de Rothmund-Thomson[3].
Manifestations cliniques
La maladie atteint deux fois plus les garçons que les filles[4]. L'âge de découverte est surtout durant l'enfance mais la maladie peut être dépistée, dans certains cas, seulement à l'âge adulte[4].
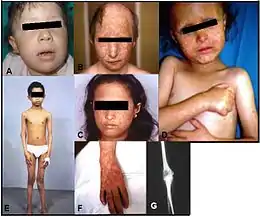
La peau est normale à la naissance, mais vers l'âge de trois à six mois se développe une poïkilodermie au niveau des joues du visage qui peut s’étendre aux bras, aux jambes et aux fesses. La poïkilodermie est une rougeur très caractéristique du SRT dont sont porteurs tous les gens atteints. La poïkilodermie — « érythème télangiectasique », en anglais on dit « rash » — est créée par une pigmentation de la peau qui varie en ces endroits, de minces vaisseaux sanguins apparents à la surface de la peau et un amincissement de la peau. Certains érythèmes comportent aussi des gonflements ou décollements épidermiques (ampoule) au niveau du visage ou des points d'appui (fesses). Ces manifestations cutanées inaugurent des signes de vieillissement prématuré incluant des troubles de la pigmentation (hypopigmentation ou hyperpigmentation), une atrophie et apparition de télangiectasies.
Les anomalies du squelette comprennent une formation anormale de l'os ou même une absence totale d'os comme le radius et d’autres déformations des os. D’autres manifestations sont des anomalies des dents et des ongles, des vomissements et des maux de ventre, des cataractes juvéniles, une petite taille, des cheveux, cils et sourcils absents ou épars et clairsemés et des cancers de la peau et des os.
Une complication courante est la survenue d'un ostéosarcome[4] ou d'autres types de cancers[5].
Traitement
Il n'existe pas de traitement de la cause de la maladie, la prise en charge se limitant à ses conséquences.
Notes et références
- (en) Kitao S, Lindor NM, Shiratori M, Furuichi Y, Shimamoto A, « Rothmund-Thomson syndrome responsible gene, RECQL4: genomic structure and products » Genomics. 1999;61:268–276
- (en) Wang LL, Gannavarapu A, Kozinetz CA et al. « Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome » J Natl Cancer Inst. 2003;95:669–674
- (en) Lisa L. Wang, Moise L. Levy, Richard A. Lewis, Murali M. Chintagumpala, Dorit Lev, Maureen Rogers et Sharon Plon « Clinical Manifestations in a Cohort of 41 Rothmund-Thomson Syndrome Patients » American journal of Medical Genetics. 2001;102:11-7.
- (en) Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, Plon SE, « Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients » Am J Med Genet. 2001;102:11–17
- (en) Simon T, Kohlhase J, Wilhelm C, Kochanek M, De Carolis B, Berthold F, « Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: Case report and literature review » Am J Med Genet A. 2010;152A:1575–1579
Liens externes
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:268400
- (en) Lisa L Wang, Sharon E Plon, Rothmund-Thomson Syndrome in GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005
 Portail de la médecine
Portail de la médecine