 ShareCompartir
ShareCompartir
Persons using assistive technology might not be able to fully access information in this file. For assistance, please send e-mail to: mmwrq@cdc.gov. Type 508 Accommodation and the title of the report in the subject line of e-mail.
Provisional CDC Guidelines for the Use and Safety Monitoring of Bedaquiline Fumarate (Sirturo) for the Treatment of Multidrug-Resistant Tuberculosis
Please note: An erratum has been published for this article. To view the erratum, please click here.
The material in this report originated in National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Jonathan Mermin, MD, Director, and the Division of Tuberculosis Elimination, Kenneth G. Castro, MD, Director.
Corresponding preparer: Sundari Mase, MD, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, CDC. Telephone: 404-639-5336; E-mail: fyy0@cdc.gov.
Summary
Multidrug-resistant tuberculosis (MDR TB) is caused by Mycobacterium tuberculosis that is resistant to at least isoniazid and rifampin, the two most effective of the four first-line TB drugs (the other two drugs being ethambutol and pyrazinamide). MDR TB includes the subcategory of extensively drug-resistant TB (XDR TB), which is MDR TB with additional resistance to any fluoroquinolone and to at least one of three injectable anti-TB drugs (i.e., kanamycin, capreomycin, or amikacin). MDR TB is difficult to cure, requiring 18–24 months of treatment after sputum culture conversion with a regimen that consists of four to six medications with toxic side effects, and carries a mortality risk greater than that of drug-susceptible TB.
Bedaquiline fumarate (Sirturo or bedaquiline) is an oral diarylquinoline. On December 28, 2012, on the basis of data from two Phase IIb trials (i.e., well-controlled trials to evaluate the efficacy and safety of drugs in patients with a disease or condition to be treated, diagnosed, or prevented), the Food and Drug Administration (FDA) approved use of bedaquiline under the provisions of the accelerated approval regulations for "serious or life-threatening illnesses" (21CFR314.500) (Cox EM. FDA accelerated approval letter to Janssen Research and Development. Available at http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/204384Orig1s000ltr.pdf).
This report provides provisional CDC guidelines for FDA-approved and unapproved, or off-label, uses of bedaquiline in certain populations, such as children, pregnant women, or persons with extrapulmonary MDR TB who were not included in the clinical trials for the drug. CDC's Division of TB Elimination developed these guidelines on the basis of expert opinion informed by data from systematic reviews and literature searches. This approach is different from the statutory standards that FDA uses when approving drugs and drug labeling. These guidelines are intended for health-care professionals who might use bedaquiline for the treatment of MDR TB for indicated and off-label uses. Aspects of these guidelines are not identical to current FDA-approved labeling for bedaquiline.
Bedaquiline should be used with clinical expert consultation as part of combination therapy (minimum four-drug treatment regimen) and administered by direct observation to adults aged ≥18 years with a diagnosis of pulmonary MDR TB (Food and Drug Administration. SIRTURO [bedaquiline] tablets label. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/204384s000lbl.pdf). Use of the drug also can be considered for individual patients in other categories (e.g., persons with extrapulmonary TB, children, pregnant women, or persons with HIV or other comorbid conditions) when treatment options are limited. However, further study is required before routine use of bedaquiline can be recommended in these populations.
A registry for persons treated with bedaquiline is being implemented by Janssen Therapeutics to track patient outcomes, adverse reactions, laboratory testing results (e.g., diagnosis, drug susceptibility, and development of drug resistance), use of concomitant medications, and presence of other comorbid conditions. Suspected adverse reactions (i.e., any adverse event for which there is a reasonable possibility that the drug caused the adverse event) and serious adverse events (i.e., any adverse event that results in an outcome such as death, hospitalization, permanent disability, or a life-threatening situation) should be reported to Janssen Therapeutics at telephone 1-800-526-7736, to FDA at telephone 1-800-332-1088 or at http://www.fda.gov/medwatch, and to CDC's Emergency Operations Center at telephone 1-770-488-7100.
Introduction
Tuberculosis (TB) is caused by the bacteria of the Mycobacterium tuberculosis complex, most commonly M. tuberculosis. TB usually is transmitted from one person to another by airborne droplet nuclei containing the bacteria. For most persons who have drug-susceptible TB, cure is achieved with a combination of first-line drugs (e.g., isoniazid [INH], rifampin [RIF], ethambutol [EMB], and pyrazinamide [PZA]) administered as a 6-month standard regimen. In contrast, multidrug-resistant tuberculosis (MDR TB), defined as TB that is caused by M. tuberculosis resistant to at least INH and RIF, generally requires 18–24 months of treatment after sputum culture conversion (SCC) with five or six drugs (e.g., susceptible first-line drugs plus an injectable agent, a fluoroquinolone, and other second-line drugs as needed) that are less effective, more toxic, and more costly than a standard first-line regimen (1,2).
MDR TB impacts communities worldwide and poses an urgent public health threat that transcends borders. In 2011, an estimated 630,000 cases of MDR TB (range: 460,000–790,000) occurred among the world's 12 million persons with prevalent cases of TB. An estimated 3.7% of persons with newly diagnosed TB and 20% of persons with previously treated TB have MDR TB. Extensively drug-resistant tuberculosis (XDR TB), defined as MDR TB with additional resistance to any fluoroquinolone and to at least one of three injectable anti-TB drugs (i.e., kanamycin, amikacin, or capreomycin), has been reported in 84 countries; on average, 9% of persons with MDR TB have additional resistance qualifying as XDR TB. India and China contribute the greatest numbers of MDR TB cases to the global burden, but the Russian Federation has the highest MDR TB rates per 100,000 population (3,4). Compared with drug-susceptible TB, MDR TB causes greater morbidity and mortality, and overall patient outcomes are worse (i.e., death, relapse [reverting to sputum culture-positive after becoming culture-negative while on treatment], treatment failure [remaining sputum culture-positive while on treatment], or disability). Mortality rates for patients being treated for MDR-TB usually exceed 10% (range: 8%–21%) (5). A recently published individual patient data meta-analysis of 9,153 patients with MDR TB yielded a mortality rate of 15% (6).
MDR TB develops when TB that is susceptible to first-line drugs is not treated adequately because of the selection of substandard treatment regimens or nonadherence with (or interruption in) treatment. Other factors (e.g., malabsorption of drugs or drug-drug interactions) also can lead to the selection of drug-resistant strains (2). Once drug-resistant TB has developed, person-to-person transmission is possible, potentially leading to outbreaks (7). MDR TB must be rapidly diagnosed and treated with an effective drug regimen to prevent further transmission of these difficult-to-cure strains of TB. One of the challenges in the treatment of MDR TB is the lack of effective, well-tolerated medications. There are very few medications and fewer classes of medications for treatment of MDR TB, and adverse drug reactions commonly necessitate discontinuing medications (2).
On December 28, 2012, the Food and Drug Administration (FDA) approved the use of bedaquiline fumarate (Sirturo or bedaquiline) as part of combination therapy (minimum four-drug therapy) administered by direct observation to adults aged ≥18 years with a diagnosis of pulmonary MDR TB when an effective treatment regimen cannot otherwise be provided (e.g., because of extensive resistance, drug intolerance, or drug-drug interactions) (8). The recommended dose of bedaquiline for the treatment of pulmonary MDR TB in adults is 400 mg administered orally once daily for 2 weeks, followed by 200 mg administered orally three times weekly, for an entire treatment duration of 24 weeks. Bedaquiline is taken with food and in combination with other anti-TB drugs. The drug is available in 100 mg tablets (9,10).
FDA approves drug products for lawful marketing for specific intended uses based on data that establish safety and efficacy, and approves labeling specific to those uses. FDA approved bedaquline as part of combination therapy to treat adults with pulmonary MDR TB when other alternatives are not available. The drug was approved under FDA's accelerated approval program, which allows the agency to approve a drug to treat a serious disease based on clinical data showing that the drug has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit to patients (11). FDA approved bedaquiline with a black box warning alerting health-care professionals to an increase in all-cause mortality and to a prolongation of the QTcF* in patients treated with bedaquiline versus placebo.
Bedaquiline, a diarylquinoline, is the first drug with a novel mechanism of action against M. tuberculosis that has been approved by FDA since 1971 (12). FDA considered this new drug under the provisions of the accelerated approval regulations for "serious or life-threatening illnesses" (21CFR314.500) (11). Bedaquiline uses adenosine 5'-triphosphate (ATP) synthase inhibition as its mechanism of action, has in vitro activity against both replicating and nonreplicating bacilli, and has bactericidal and sterilizing activity in the murine model of TB infection. No cross-resistance was found between bedaquiline and the following drugs: INH, RIF, EMB, PZA, streptomycin, amikacin or moxifloxacin. A fourfold increase in bedaquiline minimal inhibitory concentration (MIC) values (suggesting acquired resistance) has been observed in 13 of 28 patients with paired M. tuberculosis (baseline and post-baseline) isolates during clinical studies; 10 of 13 patients had isolates with matching genotypes, which is evidence against reinfection with a new M. tuberculosis strain, and, of these, nine had evidence of treatment failure or relapse. In vitro studies have demonstrated that bedaquiline has a bacteriostatic effect at low serum levels (0.3 µg/ml) that might predispose to acquired resistance (9,10).
This report provides provisional CDC guidelines for the use and safety monitoring of bedaquiline. Certain information included in these guidelines goes beyond the approved labeling for the particular products or indications in question. Clinicians should exercise judgment in management decisions modified as clinically indicated for unique patient circumstances.
Guideline Development Methods
To develop treatment guidelines for the use and safety monitoring of bedaquiline in the treatment of MDR TB, CDC's Division of TB Elimination (DTBE) conducted a literature review of published clinical trials, reviewed results of other publicly available resources (transcript and background materials from the FDA Anti-Infectives Advisory Committee Meeting of November 27, 2012, new drug application reviews, approval letter, and approved drug label) (8–11,13), and the proceedings of a CDC external consultation meeting held on January 15–16, 2013.
Published trials: A systematic literature review was performed searching PubMed/Medline with key words and search terms "bedaquiline," "multidrug resistant-tuberculosis," and "clinical trials." The search identified two publications from one double-blind, randomized, placebo-controlled Phase IIb superiority trial, C208 Stage 1, an exploratory stage that assessed 8-week bedaquiline treatment in patients with MDR TB (14,15). Phase IIa early bactericidal activity studies and review articles were excluded.
Other publicly available data: Information from the Anti-Infectives Advisory Committee Meeting Materials (9,10,13), FDA accelerated approval letter (11), and final printed label (8) were reviewed, including findings from 16 unpublished clinical pharmacology trials that evaluated the pharmacokinetics (PK) and pharmacodynamics (PD) of bedaquiline in healthy volunteers and drug interactions, and findings from two additional unpublished Phase IIb clinical trials presented to FDA at Stage 2 of drug approval by Janssen Therapeutics (C208 Stage 2, a proof-of efficacy, double-blind, randomized, placebo-controlled superiority trial separate and distinct from Stage 1, and C209, a noncomparative, single-arm, open-label trial). Both C208 Stage 1 and C208 Stage 2 had been completed, and C209 was ongoing at the time of the Advisory Committee Meeting.
Expert consultation: Following FDA approval of bedaquiline on December 28, 2012, CDC convened an ad-hoc panel of 26 external consultants†, including three FDA representatives, on January 15–16, 2013. The purpose of the meeting was for the members of the panel to provide individual expert opinion to develop provisional guidelines for the use and safety monitoring of this new drug. Each of the consultants had demonstrated TB-specific expertise in at least one of the following areas: diagnosis, treatment, prevention, nursing case management, public health programs, surveillance, epidemiology, clinical research, pulmonology, infectious diseases, pediatrics, health communication and education, migrant worker health, patient advocacy, or health economics. The external consultants and CDC staff reviewed findings from the two Phase IIb clinical trials from FDA Advisory Committee meeting materials (documents prepared by both Janssen Therapeutics and FDA) and from presentations by FDA experts, and CDC summarized the presentations and discussions of available evidence and individual expert opinions. The expert group discussed a set of questions selected by a bedaquiline workgroup comprising DTBE personnel. A summary of this discussion was prepared.
Assessment of evidence and drafting of guidelines: On the basis of the review of the literature, data from publicly available sources, and the summary of the external consultation, CDC staff drafted provisional guidelines for the use of bedaquiline. Each element of the guidelines for use of bedaquiline is rated according to the quality of the evidence. High-quality evidence is defined as data obtained from randomized controlled trials (RCTs). Low-quality evidence includes data obtained from observational studies or case series. The quality of evidence from RCTs could be downgraded to low if substantial degrees of any of the following were noted:
- bias, e.g., adequate blinding was lacking despite randomization, with substantial differences in control and intervention groups;
- indirectness, e.g., standard outcome measures were not used in the study;
- imprecise evidence, e.g., there were wide confidence intervals; or
- inconsistency and lack of evidence of reproducibility, e.g., different studies had discordant results or studies were insufficient to determine whether results are reproducible.
When evidence was lacking, a rating of "insufficient" was given to certain questions considered at the expert consultation. However, because CDC decided that provisional guidelines for these questions was important, CDC expert opinion, informed by the external expert consultation, is provided in response to those questions.
Summary of Evidence from Clinical Pharmacology Studies and Clinical Trials of Bedaquiline
Data below are presented from FDA's analysis of Janssen Therapeutics studies (9,10).
Clinical Pharmacology Studies
Results from the 16 clinical pharmacology trials (Table 1) including participants with median age 32.5 years (range: 18–68 years) indicate that peak plasma concentration and plasma exposure of bedaquiline increased approximately twofold when administered with high-fat food. Bedaquiline is highly protein-bound (>99%), is metabolized chiefly through the cytochrome P450 (CYP) system, and is excreted primarily via the feces. The mean terminal half-life (t1/2 term) of bedaquiline and its major metabolite (M2), which is four to six times less active in terms of antimycobacterial potency, is approximately 5.5 months. This long elimination phase probably reflects slow release of bedaquiline and M2 from peripheral tissues. Reproduction studies performed in rats and rabbits have revealed no evidence of harm to the fetus attributable to bedaquiline. However, no adequate and well-controlled studies have been conducted in women, and consequently bedaquiline is considered to be an FDA-assigned pregnancy category B drug (16). Whether bedaquiline or its metabolites are excreted in human milk is not known, but studies in rats have demonstrated that the drug is concentrated in breast milk. Bedaquiline distribution into the central nervous system and cerebrospinal fluid have not been evaluated. The PK of bedaquiline in pediatric patients (aged 0–17 years) and elderly persons (aged ≥65years) has not been evaluated. Black patients have a clearance of bedaquiline that is 52% higher than that of patients who were classified as non-Hispanic white, Asian, Hispanic, or other. This could result in lower systemic exposure in black patients than in patients in these other racial/ethnic categories.
CYP3A4 is the major CYP isoenzyme involved in the metabolism of bedaquiline. Drug-drug interactions occurred with CYP3A4 inducers (e.g., RIF reduced bedaquiline exposure by approximately 50%) and CYP3A4 inhibitors (e.g., ketoconazole increased bedaquiline exposure by approximately 22%). No significant PK interactions were observed with the anti-TB drugs INH, PZA, EMB, kanamycin, ofloxacin, or cycloserine, or with the antiretroviral drug nevirapine; however, lopinavir/ritonavir increased bedaquiline exposure by 22%. Additional information on drug-drug interactions is available in the bedaquiline label (8). (For additional information on use of bedaquiline in patients with renal and hepatic impairment, see Precautions and Monitoring for Adverse Events.)
Efficacy Studies
Efficacy of bedaquiline was assessed in three Phase IIb studies: Studies C208 Stage 1, C208 Stage 2 (both randomized placebo-controlled trials), and C209 (a noncomparative, single-arm open-label trial) (Table 2). Study C208 evaluated efficacy in patients with newly diagnosed MDR TB in two completely separate studies conducted consecutively: an exploratory study of 8 weeks of bedaquiline with a standard five-drug background regimen consisting of ethionamide, kanamycin, PZA, ofloxacin, and cycloserine/terizidone (Stage 1) and a proof-of-efficacy study of 24 weeks of bedaquiline with the same standard background regimen (Stage 2). Study C209 assessed efficacy in patients with MDR TB with varying degrees of additional resistance who failed previous therapy and were on individualized background regimens tailored either to the susceptibility pattern of their M. tuberculosis isolate or to their treatment history. The primary endpoint in all studies was time to SCC, defined as two consecutive cultures from sputum that were negative for M. tuberculosis in a modified intention-to-treat analysis (mITT) (Tables 2–4).
In Study C209, patients with laboratory-confirmed pulmonary MDR TB were treated with bedaquiline plus background regimen. The median time to SCC was 57 days (Figure 1) and consistent with both C208 Stage 1 and Stage 2 results (data not presented).
Safety Studies
Safety in Healthy Volunteers
From 2005 to 2012, Janssen Therapeutics conducted 11 Phase I studies to evaluate bedaquiline. Analysis of pooled safety data from eight Phase 1 studies (five single-dose studies and three multiple-dose studies) enrolling 189 healthy adult patients who received at least 1 dose of bedaquiline support the safety and tolerability of bedaquiline administered either alone or with selected other medications. Of the 57 multiple-dose patients, 47 received bedaquiline for at least 14 days, and 37 received bedaquiline with other medication (INH and PZA: 22 patients; ketoconazole: 15 patients). Suspected adverse reactions were solicited actively at the time of drug administration. No deaths or serious adverse events were reported. No patient exposed to bedaquiline, either alone or in combination with other drugs, had a QTcF >500 milliseconds (ms). No subject discontinued treatment because of QTcF prolongation.
Safety in Patients with Drug-Susceptible Tuberculosis Treated for 7 days
Trial C202 was a proof-of-principle, open-label, active-controlled, randomized Phase IIa trial in 45 patients with drug-susceptible TB. Patients were randomized to five groups receiving 7 days of monotherapy with bedaquiline (25 mg, 100 mg, or 400 mg) or INH (300 mg) or RIF (600 mg). Of 75 patients enrolled, 45 received bedaquiline. No deaths occurred from an adverse drug reaction that started during the 7-day investigational study phase (bedaquiline, INH, or RIF) treatment period. Two patients (both of whom were in the bedaquiline 400 mg group) died during the follow-up period, after initiation of a treatment regimen for their drug-susceptible TB consisting of INH, RIF, EMB, and PZA; neither death was attributable to bedaquiline. The rate of serious adverse events was balanced in both treatment groups. A greater increase in QTcF was noted at 5 hours postdose compared with baseline in patients who received 400 mg (median increase: 24.5 ms) compared with patients who received 100 mg (median increase: 10.1 ms) or 25 mg (no increase) of bedaquiline or 300 mg of INH (median increase: 15.8 ms) or 600 mg of RIF (median increase: 13.1 ms).
Safety in Patients with Drug-Resistant TB
The safety and tolerability of bedaquiline for the treatment of pulmonary MDR TB in adults as part of combination therapy was assessed by pooling of data from C208 Stage 1 and Stage 2 (207 TB patients, 102 of whom received bedaquiline: 23 for 8 weeks and 79 for 24 weeks) and C209 (233 TB patients, all of whom received bedaquiline for 24 weeks) to increase the likelihood of detecting infrequent adverse events attributable to the higher number of patients per pooled treatment group and to increase the sample size for subgroup analyses (Table 5). Excluding 233 patients treated in C209, comparative safety (to placebo plus background regimen) data were available for 102 of these 335 patients.
The most frequent suspected adverse reactions (>20.0% of patients) during treatment with bedaquiline in the controlled trials (C208 Stages 1 and 2) were nausea (35.3%), arthralgia (29.4%), headache (23.5%), hyperuricemia (22.5%), and vomiting (20.6%). The incidence of these adverse drug reactions in the bedaquiline arm was similar to the placebo arm except for headache (in 23.5% and 11.4% of patients, respectively), nausea (35.3% and 25.7%, respectively), and arthralgia (29.4% and 20.0%, respectively).
Over the 24-week study drug-treatment period in C208 Stage 2, hepatic-related adverse drug reactions and increase in QTcF were observed more commonly in the bedaquiline arm compared with the placebo arm (8). Two patients treated with bedaquiline had serious hepatic adverse reactions, one of whom had elevation in total bilirubin (peak 52 µmol/L at 24 weeks). One (1.3%) patient in the bedaquiline arm had a QTcF value of >500 ms, compared with none in the placebo arm; one patient each from the bedaquiline and placebo arms developed QTcF values between 480 and 500 ms; and QTcF values between 450 and 480 ms were observed in 26.6% and 8.6% of patients in the bedaquiline and placebo arm, respectively. Seven (9.1%) patients in the bedaquiline arm had a >60 ms increase from baseline in QTcF compared with two (2.5%) patients in the placebo arm. Fifty-six percent of patients in the bedaquiline arm and 31.6% of patients in the placebo arm experienced increases in QTcF between 30 to 60 ms. No episodes of torsade de pointes (a polymorphic ventricular tachycardia that can degenerate into ventricular fibrillation and is associated with QTcF prolongation) occurred in patients with QTcF prolongation. The magnitude of QTcF prolongation over baseline increased in the first 18 weeks of bedaquiline treatment, remained relatively stable until week 24, and then gradually decreased (Figure 2). The magnitude of QTcF prolongation was greater in patients treated concomitantly with clofazimine.
Deaths Among Clinical Trial Participants
A total of 36 deaths were reported during the entire clinical development program of bedaquiline: 30 in the bedaquiline group and six in the placebo group (Table 6). In C208 Stage 2, nine of the 10 deaths in the bedaquiline arm, and two of the four deaths in the placebo arm occurred within the 120-week cutoff period for the efficacy analysis. For five of the nine bedaquiline-treated patients who died and both of the placebo patients who died, the cause of death was TB; no clear cause of death related to bedaquiline toxicity was identified. Detailed analysis of baseline characteristics and risk factors for poor treatment outcomes did not reveal relevant imbalances to explain the increased mortality observed in the bedaquiline arm. There was no relationship between bedaquiline serum levels or QTcF >500 ms and survival outcome. Nevertheless, because of the excess deaths and QT interval effects in the treatment group, the prescribing information included the following black box warnings:
- An increased risk for death was observed in the Sirturo treatment group (9/79; 11.4%) compared with the placebo treatment group (2/81; 2.5%) in one placebo-controlled trial. Only use Sirturo when an effective treatment regimen cannot otherwise be provided;
- QTcF prolongation can occur with Sirturo. Use with drugs that prolong the QT interval might cause additive QTcF prolongation.
CDC Provisional Guidelines for the Use of Bedaquiline
Bedaquiline may be used for 24 weeks of treatment in adults with laboratory-confirmed pulmonary MDR TB (TB with an isolate showing genotypic or phenotypic resistance to both INH and RIF) when an effective treatment regimen cannot otherwise be provided.
Quality of evidence: low.
Expert opinion: The possible benefits of using bedaquiline outweigh the potential risk.
Evidence basis and rationale: One published RCT (C208 Stage 1) showed bedaquiline used for 8 weeks was superior to placebo at achieving an earlier time to SCC and a statistically significant increase in SCC rate at 8 weeks in patients with pulmonary MDR TB who were treated with optimized background regimen. The gold standard for outcomes of MDR TB treatment is persistent negative sputum culture 2 years after treatment completion, and time to SCC and 2-month SCC rate are imperfect surrogate markers. Only one RCT noted above evaluated a 8-week bedaquiline course and reproducibility of results cannot be determined. Therefore, quality of evidence was downgraded from high to low. However, FDA reviewed data from two additional unpublished studies that supported efficacy using the endpoint of SCC: an RCT demonstrating superiority of a 24-week bedaquiline course added to background regimen in both time to SCC and 24 week SCC rate (C208 Stage 2) and a single arm trial in patients who achieved similar time to SCC as that shown in both C208 Stage 1 and Stage 2, despite having failed previous therapy (C209) (9). The higher mortality rate associated with MDR TB when compared with drug-sensitive TB supports consideration of bedaquiline use in patients for whom an effective regimen cannot otherwise be provided, despite potential toxicity concerns described above.
Bedaquiline may be used on a case-by-case basis in children, HIV-infected persons, pregnant women, persons with extrapulmonary MDR TB, and patients with comorbid conditions on concomitant medications when an effective treatment regimen cannot otherwise be provided.
Quality of evidence: insufficient.
Expert opinion: The possible benefits of using bedaquiline outweigh the potential risk.
Evidence basis and rationale: The effectiveness and safety of bedaquiline for the treatment of MDR TB have not been studied adequately in these populations to provide general guidance for or against its use. Because MDR TB has a high mortality rate and treatment options are limited, its use might be a reasonable option for providers to consider in treating certain patients in the groups listed above.
Bedaquiline may be used on a case-by-case basis for durations longer than 24 weeks when an effective treatment regimen cannot be provided otherwise.
Quality of evidence: insufficient.
Expert opinion: The possible benefits of using bedaquiline outweigh the potential risk.
Evidence basis and rationale: The effectiveness and safety of bedaquiline have not been studied beyond a duration of 24 weeks. Therefore, general guidance cannot be provided for or against its use for durations beyond 24 weeks. Because MDR TB has a high mortality rate and treatment options are limited, bedaquiline use beyond 24 weeks might be justified in some patients.
Additional Considerations
Dosing and Administration
- The recommended dosage for bedaquiline is 400 mg once daily orally for 2 weeks, followed by 200 mg three times a week for 22 weeks taken orally with food in order to maximize absorption.
- If a dose of bedaquiline is missed during the first 2 weeks of treatment, patients should not make up the missed dose but should continue the usual dosing schedule. From Week 3 onward, if a 200 mg dose is missed, patients should take the missed dose as soon as possible, and then resume the three-times-a-week regimen with established intervals (i.e., days of the week).
- Bedaquiline never should be used as a single drug and should be used only in combination with at least three other drugs (i.e., for a four-drug regimen) to which the patient's MDR TB isolate has been shown to be susceptible in vitro. If in vitro testing results are unavailable, treatment may be initiated with bedaquiline in combination with at least four other drugs (i.e., for a five-drug regimen) to which the patient's MDR TB isolate is likely to be susceptible (1,8).
- The use of bedaquiline with rifamycins or other drugs that induce or suppress CYP3A4 should be avoided, unless the anticipated benefits outweigh the risks associated with inadequate treatment of MDR TB; if bedaquiline is given with these drugs, monitoring of serum drug levels should be performed to ensure adequate therapy and minimize the risk for acquired drug resistance. Providers are encouraged to contact CDC for technical assistance and referral for monitoring serum drug levels if necessary.
- Because bedaquiline has an extremely long terminal half-life (4–5 months), acquired resistance might occur when bedaquiline is the sole effective anti-TB drug in circulation. Prescribers should consider discontinuation of bedaquiline 4–5 months before scheduled termination of other drugs in the regimen to reduce or avoid an extended period of exposure to low levels of bedaquiline as a single drug.
- Bedaquiline should be administered only by directly observed therapy (DOT) and with case management strategies. Such strategies might include the use of incentives and enablers (e.g., food certificates, bus passes, cash, or housing) to ensure adherence to the treatment regimen.
- Patients should be advised that nonadherence to a treatment regimen could result in treatment failure, relapse, or acquired resistance. An evaluation for the development of resistance to the anti-TB regimen (including repeat drug susceptibility testing, if indicated) is recommended for patients with treatment failure or relapse.
Monitoring
- Persons receiving bedaquiline should be monitored weekly for nausea, headache, hemoptysis, chest pain, arthralgia and rash, and treatment should be modified as clinically indicated.
- Monitoring for other suspected adverse reactions should be tailored to side effects specific to other drugs in the background regimen and the potential for drug interactions with bedaquiline.
- Bedaquiline use in the treatment of pulmonary MDR TB should be accompanied by microbiologic monitoring with one sputum specimen submitted for culture monthly throughout and at end of treatment, even after conversion to negative culture, which is consistent with the standard approach to treatment and care of patients with MDR TB in the United States. Any monthly specimen that grows M. tuberculosis, including one before treatment initiation with bedaquiline, should be referred to a laboratory for surveillance of bedaquiline resistance in consultation with the state public health laboratory (1,17–19). CDC will assist in identifying a laboratory that can perform bedaquiline susceptibility testing for this purpose.
Precautions and Monitoring for Adverse Events
- Hepatotoxicity
— Persons receiving bedaquiline should be advised to avoid alcohol and other hepatotoxic drugs and should be monitored closely as noted below (8).
— Bedaquiline can be administered to patients with mild to moderate hepatic impairment (Child-Pugh A or B), but should be avoided in patients with severe hepatic impairment (Child-Pugh C) (20).
— Aspartate aminotransferase (AST), alanine aminotransferase (ALT), bilirubin, and alkaline phosphatase should be monitored at baseline, monthly, and if symptomatic; more frequent monitoring should be considered if administered with other hepatotoxic medications or in those with underlying liver disease.
— An increase of serum aminotransferases to more than three times the upper limit of normal (ULN) should be followed by repeat testing within 48 hours. American Thoracic Society (ATS)/CDC/Infectious Diseases Society of America (IDSA) treatment guidelines for management of hepatotoxicity should be followed (1). Testing for viral hepatitis should be performed and other hepatotoxic medications discontinued.
— Evidence of new or worsening liver dysfunction (including clinically significant elevation of aminotransferases or bilirubin or the appearance of symptoms such as fatigue, anorexia, nausea, jaundice, dark urine, liver tenderness, or hepatomegaly) should prompt additional evaluation by the prescriber.
— Discontinuation of bedaquiline should be considered if the following laboratory abnormalities are present:
- aminotransferase elevations accompanied by total bilirubin elevation more than two times ULN,
- aminotransferase elevations more than eight times ULN, and
- aminotransferase elevations that persist beyond 2 weeks.
- Renal impairment
— Bedaquiline is minimally excreted by the kidneys and does not require dosage adjustment in patients with mild to moderate renal impairment (not requiring dialysis), and should be administered with caution in patients with severe renal impairment requiring dialysis.
— Obtaining serum drug levels in patients with renal impairment should be considered. - Cardiac toxicity
— Persons receiving bedaquiline should be monitored for signs of cardiac toxicity closely with an ECG.,
— Baseline ECG should be obtained and repeated at least 2, 12, and 24 weeks after starting treatment
— Serum potassium, calcium, and magnesium should be obtained at baseline and whenever clinically indicated, especially if QTcF prolongation is detected. Any abnormalities that are detected should be addressed, and electrolytes should be monitored until fully corrected.
— The concurrent use of other drugs known also to cause QTcF prolongation could be associated with increased risk of cardiotoxicity when patients are receiving bedaquiline. Persons who receive bedaquiline should be monitored weekly with an ECG if any of the following circumstances are present:
- they receive other QTcF prolonging drugs, including fluoroquinolones, macrolide antibacterial drugs, and the antimycobacterial drug clofazimine;
- they have a history of torsade de pointes, congenital long QT syndrome, hypothyroidism and bradyarrhythmias, or uncompensated heart failure; or
- they have serum calcium, magnesium, or potassium levels below the lower limits of normal.
- clinically significant ventricular arrhythmia, or
- a QTcF of >500 ms (confirmed by repeat ECG).
— After bedaquiline is discontinued for cardiotoxicity, ECGs should be monitored frequently to confirm that QTcF has returned to baseline.
— If syncope occurs, an ECG should be obtained to evaluate for QTcF prolongation.
Training, Education, and Medical Consultation
- Training and education for bedaquiline administration, adverse event monitoring, clinical response monitoring, and reporting should be provided for public health programs/professionals, TB physicians, and others who will be involved in the care and management of patients receiving bedaquiline.
- Treatment of MDR TB should be provided in consultation with an expert in the management of MDR TB.
Registry of Patients Treated With Bedaquiline
Per postmarketing surveillance requirements (11), all persons started on bedaquiline should be enrolled in a patient registry. This registry should facilitate an expanded and standardized monitoring system to track systematically such variables as serious adverse events, less serious side effects, early and late patient outcomes (culture conversion, relapse, completion of treatment, and cure), laboratory data including collection and testing of patient isolates at the start of and during treatment, interaction with other drugs, use in multidrug regimens, drug administration practices, use in the operational setting, feasibility of implementation and cost effectiveness, drug distribution mechanisms and process, and drug utilization data. Janssen Therapeutics will maintain this registry and collect data prospectively on all patients started on bedaquiline through December 2018 with a final report anticipated in August 2019. CDC, in conjunction with Janssen Therapeutics, will provide guidance on the implementation plan for bedaquiline use (process for distribution through an identified point of contact for each of the 68 DTBE-funded jurisdictions in the United States).
Safety Reporting
Suspected adverse reactions (i.e., any adverse event for which there is a reasonable possibility that the drug caused the adverse event) and serious adverse events (i.e., any adverse event that results in an outcome such as death, hospitalization, permanent disability, or a life-threatening situation) should be reported to Janssen Therapeutics at telephone 1-800-526-7736, to FDA at telephone 1-800-332-1088 or at http://www.fda.gov/medwatch, and to CDC's Emergency Operations Center at telephone 1-770-488-7100.
Planned Studies and Updates to CDC
Provisional Guidelines
Janssen Therapeutics is required to conduct a double-blind placebo-controlled multicenter Phase III RCT in patients with sputum smear-positive pulmonary MDR TB as part of the conditions associated with accelerated approval under 21CFR314.500 (11). Additional requirements include, but are not limited to, a study to define the quality control ranges of bedaquiline for MDR TB isolates using standard agar proportion and MIC methods, and a clinical trial to identify any unexpected serious risk of increased drug levels of bedaquiline in HIV patients co-infected with M. tuberculosis. CDC will revise these provisional CDC guidelines as needed when those data become available.
References
- CDC. Treatment of tuberculosis. MMWR 2003;52(No. RR-11).
- Francis J. Curry National Tuberculosis Center, California Department of Health Service. Drug-resistant tuberculosis: a survival guide for clinicians. 2nd ed. San Francisco, CA: Francis J. Curry National Tuberculosis Center, California Department of Public Health; 2011.
- World Health Organization. Global tuberculosis report 2012. Geneva, Switzerland: World Health Organization; 2012.
- World Health Organization. Multidrug and extensively drug-resistant tuberculosis (M/XDR-TB). 2010 global report on surveillance and response. Geneva, Switzerland: World Health Organization; 2010.
- Wells CD. Tuberculosis among HIV-infected and other immunocompromised hosts: epidemiology, diagnosis, and strategies for management. Curr Infect Dis Rep 2010;12:192–7
- Johnston JC, Shahidi NC, Sadatsafavi M, Fitzgerald JM. Treatment outcomes of multidrug-resistant tuberculosis: a systematic review and meta-analysis. PLoS ONE 4[9]:e6914.doi:10.1371/journal.pone.0006914
- Gandhi NR, Moll A, Sturm AW, et al. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 2006;368:1575–80.
- Food and Drug Administration. SIRTURO (bedaquiline) tablets label. Washington, DC: Food and Drug Administration; 2012. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/204384s000lbl.pdf.
- Food and Drug Administration. Briefing package: Sirturo. Washington, DC: Food and Drug Administration; 2012. Available at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM329258.pdf.
- Food and Drug Administration. Briefing package: TMC207 (bedaquiline). Treatment of patients with MDR-TB. Washington, DC: Food and Drug Administration; 2012. Available at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM329260.pdf.
- Cox EM. FDA accelerated approval letter to Janssen Research and Development. Washington, DC: Food and Drug Administration; 2012. Available at http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/204384Orig1s000ltr.pdf.
- Sensi P. History of the development of rifampicin. Rev Infect Dis 1983;5(Suppl 3):S402–6.
- Food and Drug Administration. Drugs for the treatment of tuberculosis, including drug resistant tuberculosis. Washington, DC: Food and Drug Administration; 2012. Available at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM188373.pdf.
- Diacon AH, Pym A, Grobusch M, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 2009;360:2397–405 10.1056/NEJMoa0808427.
- Diacon AH, Donald PR, Pym A, et al. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob Agents Chemother 2012;56:3271–6 10.1128/AAC.06126-11.
- Food and Drug Administration. FDA Pregnancy categories. Washington, DC: Food and Drug Administration; 2004. Available at http://depts.washington.edu/druginfo/Formulary/Pregnancy.pdf.
- Kurbatova EV, Gammino VM, Bayona J, et al. Predictors of sputum culture conversion among patients treated for multidrug‐resistant tuberculosis. Int J Tuberc Lung Dis 2012;16:1335–43.
- Kurbatova EV, Gammino VM, Bayona J, et al. Frequency and type of microbiological monitoring of multidrug-resistant tuberculosis treatment. Int J Tuberc Lung Dis 2011;15:1553–5.
- CDC. Tuberculosis laboratory information. Atlanta, GA: US Department of Health and Human Services, CDC; 2013. Available at http://www.cdc.gov/tb/topic/laboratory/default.htm.
- Child CG, Turcotte JG. Surgery and portal hypertension. In: Child CG, ed. The liver and portal hypertension. Philadelphia, PA: Saunders; 1964:50–64.
* The QT interval is a measure of the time between the start of the Q wave and the end of the T wave in the heart's electrical cycle. A lengthened QT interval is a biomarker for ventricular tachyarrhythmias and a risk factor for sudden death. The QT interval is dependent on the heart rate and may be corrected by calculation to improve the detection of patients at increased risk of ventricular arrhythmia. One of several calculation correction formulas focuses on the QT interval divided by cube-root of RR (QTcF), where RR is the interval from the onset of one QRS complex (the graphical deflections seen on an electrocardiogram [ECG] that correspond to the depolarization of the right and left ventricle with each heart beat) to the onset of the next QRS complex, measured in milliseconds.
† A list of the members of the panel appears on page 12. All members of the panel submitted a signed form declaring that they had no conflicts of interest or competing interests.
FIGURE 1. Time to sputum culture conversion (SCC) in patients treated with bedaquiline who failed previous therapy for drug-resistant pulmonary tuberculosis
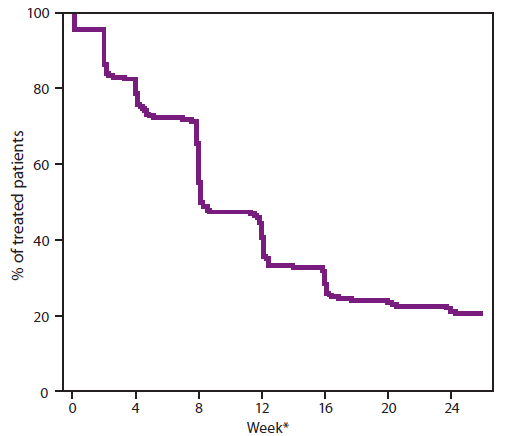
Source: Adapted from Food and Drug Administration clinical pharmacology review (9).
* Time to SCC (two consecutive cultures from sputum samples that were negative for Mycobacterium tuberculosis) in weeks.
Alternate Text: The figure above shows the time to sputum culture conversion (two consecutive cultures from sputum samples that were negative for Mycobacterium tuberculosis) in weeks. By week 24, the percentage of treated patients who not yet achieved sputum culture conversion (SCC) declined to 20%, whereas 80% had achieved SCC.
|
Study (Stage) |
Design |
No. of deaths |
|||
|---|---|---|---|---|---|
|
Bedaquiline arm |
Control arm |
||||
|
No. |
(%) |
No. |
(%) |
||
|
C202 |
Randomized, open-label, dose-ranging early bactericidal study using INH or RIF in control arm |
2/45 |
(4.4) |
0 |
0 |
|
C208 (Stage 1) |
Double-blind, randomized, placebo-controlled superiority trial |
2/23 |
(8.7) |
2/124 |
(8.3) |
|
C208 (Stage 2) |
Double-blind, randomized, placebo-controlled superiority trial |
10/79 |
(12.6) |
4/81 |
(4.9) |
|
C209 |
Noncomparative, single-arm, open-label trial |
16/233 |
(6.9) |
No |
No |
|
Source: Adapted from Food and Drug Administration clinical pharmacology review (9). Abbreviations: INH = isoniazid; RIF = rifampin. * Patients in the mortality analysis were followed for up to 6 months from the last recorded visit, as specified in the study safety procedures. |
|||||
FIGURE 2. Mean changes from baseline in QTcF* over time among patients treated with bedaquiline plus background regimen† (BR) versus placebo plus BR — Study C208 (Stage 2)
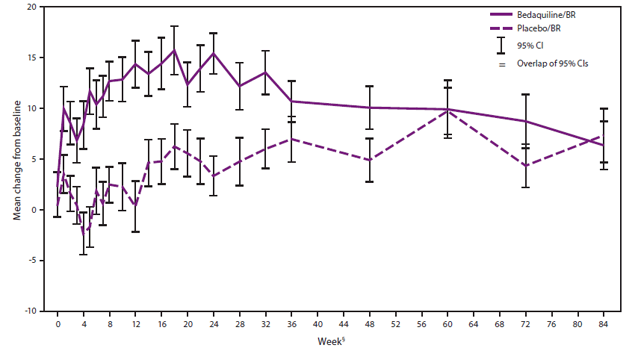
Abbreviation: 95% CI = 95% confidence interval.
Source: Food and Drug Administration primary clinical review.
* The QT interval is a measure of the time between the start of the Q wave and the end of the T wave in the heart's electrical cycle. A lengthened QT interval is a biomarker for ventricular tachyarrhythmias and a risk factor for sudden death. The QT interval is dependent on the heart rate and may be corrected by calculation to improve the detection of patients at increased risk of ventricular arrhythmia. One of several calculation correction formulas focuses on the QT interval divided by cube-root of RR (QTcF), where RR is the interval from the onset of one QRS complex (the graphical deflections seen on an electrocardiogram (ECG) that correspond to the depolarization of the right and left ventricle with each heart beat) to the onset of the next QRS complex, measured in milliseconds.
† Ethionamide, kanamycin, pyrazinamide, ofloxacin, and cycloserine/terizidone.
§ Time to smear culture conversion (two consecutive cultures from sputum samples that were negative for Mycobacterium tuberculosis) in weeks.
Alternate Text: The figure above shows the mean changes from baseline in QTcF over time among patients treated with bedaquiline plus background regimen (BR, i.e., ethionamide, kanamycin, pyrazinamide, ofloxacin, and cycloserine/terizidone) versus placebo plus BR for Study C208 (Stage 2). The QT interval is a measure of the time between the start of the Q wave and the end of the T wave in the heart's electrical cycle. A lengthened QT interval is a biomarker for ventricular tachyarrhythmias and a risk factor for sudden death. The QT interval is dependent on the heart rate and may be corrected by calculation to improve the detection of patients at increased risk of ventricular arrhythmia. One of several calculation correction formulas focuses on the QT interval divided by cube-root of RR (QTcF), where RR is the interval from the onset of one QRS complex (the graphical deflections seen on an electrocardiogram (ECG) that correspond to the depolarization of the right and left ventricle with each heart beat) to the onset of the next QRS complex, measured in milliseconds.
Bedaquiline Expert Consultants Panel
Chair: Sundari Mase, MD, CDC, Atlanta, Georgia.
Members: Tim Aksamit, MD, Mayo Clinic, Rochester, Minnesota; David Ashkin, MD, Florida Department of Health, Delray Beach, Florida; Pennan Barry, MD, University of California, San Francisco, California; Robert Belknap, MD, Denver Public Health and University of Colorado, Denver, Colorado; Henry M. Blumberg, MD, Emory University School of Medicine, Atlanta, Georgia; Dakshina M. Chilukuri, PhD, Food and Drug Administration, Silver Spring, Maryland; Edward Cox, MD, Food and Drug Administration, Silver Spring, Maryland; Charles L. Daley, MD, National Jewish Health, Denver, Colorado; Jennifer M. Flood, MD, California Department of Health, Berkeley, California; Fred M. Gordin, MD, Veterans Administration Medical Center, Washington, District of Columbia; Carol D. Hamilton, MD, Global Health, Population and Nutrition, Durham, North Carolina; Mark Harrington, Treatment Action Group, New York City, New York; Alfred Lardizabal, MD, New Jersey Medical School Global TB Institute, Newark, New Jersey; Christian Lienhardt, PhD, World Health Organization, Geneva, Switzerland; Masa Narita, MD, Seattle & King County TB Control Program, Seattle, Washington; Eileen E. Navarro Almario, MD, Food and Drug Administration, Silver Spring, Maryland; Diana M. Nilsen, MD, New York City Department of Health and Mental Hygiene, New York City, New York; Charles Peloquin, PharmD, FCCP, University of Florida College of Pharmacy, Gainesville, Florida; Ariel R. Porcalla, MD, Food and Drug Administration, Silver Spring, Maryland; Susan Ray, MD, Emory University School of Medicine, Atlanta, Georgia; Gisela Schecter, MD, California Department of Public Health, Richmond, California; Barbara J. Seaworth, MD, Heartland National TB Center, San Antonio, Texas; Jeffrey R. Starke, MD, Baylor College of Medicine, Houston, Texas; James Sunstrum, MD, Michigan Department of Community Health, Westland, Michigan; Jon Warkentin, MD, Tennessee Department of Health, Nashville, Tennessee; John W. Wilson, MD, Mayo Clinic, Rochester, Minnesota.
Use of trade names and commercial sources is for identification only and does not imply endorsement by the U.S. Department of
Health and Human Services.
References to non-CDC sites on the Internet are
provided as a service to MMWR readers and do not constitute or imply
endorsement of these organizations or their programs by CDC or the U.S.
Department of Health and Human Services. CDC is not responsible for the content
of pages found at these sites. URL addresses listed in MMWR were current as of
the date of publication.
All MMWR HTML versions of articles are electronic conversions from typeset documents.
This conversion might result in character translation or format errors in the HTML version.
Users are referred to the electronic PDF version (http://www.cdc.gov/mmwr)
and/or the original MMWR paper copy for printable versions of official text, figures, and tables.
An original paper copy of this issue can be obtained from the Superintendent of Documents, U.S.
Government Printing Office (GPO), Washington, DC 20402-9371;
telephone: (202) 512-1800. Contact GPO for current prices.
**Questions or messages regarding errors in formatting should be addressed to
mmwrq@cdc.gov.
