

Chapter 22: Laboratory Support for Surveillance of Vaccine-Preventable Diseases
 ShareCompartir
ShareCompartir
Manual for the Surveillance of Vaccine-Preventable Diseases
Printer friendly version [28 pages]
Authors: Sandra W. Roush, MT, MPH; Bernard Beall, PhD; Pam Cassiday, MS; Jon Gentsch, PhD; Joe Icenogle, PhD; Leonard Mayer, PhD; Steven M. Oberste, PhD; Daniel C. Payne, PhD, MSPH; Paul Rota, PhD; D. Scott Schmid, PhD; Michael Shaw, PhD; Maria Lucia Tondella, PhD; Annemarie Wasley, ScD
Surveillance of Vaccine-Preventable Diseases
Surveillance for vaccine-preventable diseases (VPDs) requires the close collaboration of clinicians, public health professionals, and laboratorians. Public health surveillance relies on both clinical and laboratory reports of VPDs; therefore, appropriate specimen collection, transport, and laboratory testing are essential. This chapter provides guidelines on specimen collection for each VPD and interpretation of laboratory results.
Each public health professional dealing with vaccine-preventable diseases should identify sources of laboratory support for his or her clinical and public health practice. Table 1 lists appropriate tests for VPDs and provides names and contact information for laboratories and laboratory personnel. In addition to the guidelines presented in this chapter, state health department personnel can provide additional guidance on specimen collection, transport, and other related information.
Table 1. Contact persons for VPD surveillance laboratory support
| Disease | Test name | Lab Name | Lab contact/Phone |
|---|---|---|---|
| Diphtheria |
|
CDC Pertussis and Diphtheria Laboratory | Dr. M. Lucia Tondella mlt5@cdc.gov 404-639-1239 or Ms. Pam Cassiday pxc1@cdc.gov 404-639-1231 FAX: 404-639-4421 |
| Haemophilus influenzae |
|
CDC Meningitis Laboratory |
Dr. Leonard Mayer FAX: 404-639-4421 |
| Hepatitis A | Hepatitis Reference Laboratory |
Ruth Jiles FAX: 404-639-1563 |
|
| Hepatitis B | Hepatitis Reference Laboratory |
Ruth Jiles FAX: 404-639-1563 |
|
| Influenza |
|
CDC Influenza Surveillance and Diagnosis Laboratory |
Dr. Michael Shaw FAX: 404-639-2350 |
| Measles |
|
CDC MMR & Herpes Virus Laboratory |
Dr. Paul Rota FAX: 404-639-4187 |
| Meningococcal disease |
|
CDC Meningitis Laboratory |
Dr. Leonard Mayer FAX: 404-639-4421 |
| Mumps |
|
CDC MMR & Herpes Virus Laboratory |
Dr. Paul Rota FAX: 404-639-4187 |
| Pertussis |
|
CDC Pertussis and Diphtheria Laboratory |
Dr. M. Lucia Tondella FAX: 404-639-4421 |
| Pneumococcal disease |
|
CDC Streptococcus Laboratory |
Dr. Bernard Beall FAX: 404-639-2070 |
| Poliomyelitis |
|
CDC Polio/Picornavirus Laboratory |
Dr. Steve Oberste FAX: 404-639-4011 |
| Rotavirus |
|
CDC Gastroenteritis Virus Laboratory |
Dr Jon Gentsch FAX: 404-639-3645 |
| Rubella |
|
CDC MMR & Herpes Virus Laboratory |
Dr. Joe Icenogle FAX: 404-639-1516 |
| Congenital rubella syndrome |
|
CDC MMR & Herpes Virus Laboratory |
Dr. Joe Icenogle FAX: 404-639-1516 |
| Varicella |
|
National VZV Laboratory |
Dr. Scott Schmid FAX: 404-639-4056 |
*Note for Pneumococcal Serotyping (conventional or PCR-based): Provide typing of isolates of S. pneumoniae only in the setting of an outbreak. PCR-based serotyping can be performed using commercially available supplies.
General Guidelines for Specimen Collection and Laboratory Testing
Specimen collection and shipping are important steps in obtaining laboratory diagnosis or confirmation for VPDs. Guidelines have been published for specimen collection and handling for viral and microbiologic agents.[1-3] Information is also available on using CDC laboratories as support for reference and disease surveillance;[4, 5] this includes the form required for submitting specimens to CDC (See Appendix 23, Form # CDC 0.5034) and information on general requirements for shipment of etiologic agents (Appendix 24 [4 pages]). Although written to guide specimen submission to CDC, this information may be applicable to the submission of specimens to other laboratories.
Disease-specific Guidelines for Specimen Collection and Laboratory Testing
This chapter provides a quick reference summary of the laboratory information from Chapters 1-17 of this manual. Table 2 lists confirmatory and other useful tests for surveillance of vaccine-preventable diseases.
Table 2. Confirmatory and other useful tests for the surveillance of vaccine-preventable diseases
| Disease | Confirmatory tests | Other useful tests |
|---|---|---|
| Diphtheria | Culture Toxigenicity testing |
PCR Serology (antibodies to diphtheria toxin) |
| Haemophilus influenzae | Culture | Serotyping (identification of capsular type of encapsulated strains) Antigen detection Subtyping |
| Hepatitis A | IgM anti-HAV (positive) | Total anti-HAV (marker of immunity) PCR |
| Hepatitis B | IgM anti-HBc (acute infection) HBsAg (acute or chronic infection)* |
Anti-HBs (marker of immunity) Total anti-HBc (marker of past or present infection) |
| Influenza | Culture Antigen detection (EIA, IFA, EM) Serology PCR |
|
| Measles | IgM Paired sera for IgG |
Culture (for molecular epi) PCR |
| Meningococcal disease | Culture | Serogroup-specific PCR Slide agglutination serogrouping PCR |
| Mumps | Culture IgM IgG |
IgG (for immunity testing) |
| Pertussis | Culture PCR |
Serology |
| Pneumococcal disease | Culture PCR |
Antibiotic resistance - serotyping - PCR deduction of serotypes - strain identification (MLST,PFGE) |
| Poliomyelitis | Culture-from stool, pharynx, or CSF |
Intratypic differentiation (wild vs. vaccine type) Paired serology CSF analysis |
| Rotavirus | Culture Paired serology |
Nucleic acid electrophoresis PCR genotyping |
| Rubella | Paired sera for IgG IgM Culture |
PCR |
| Tetanus | There are no lab findings characteristic of tetanus | Serology (for immunity testing) |
| Varicella | Culture Serology |
Viral typing/strain identification DFA |
* Confirmation of HBsAg positive results by HBsAg neutralization assay should be performed as specified in test package insert.
Table 3 summarizes specimen collection procedures for laboratory testing. Because some specimens require different handling procedures, be sure to check with the diagnostic laboratory prior to shipping. When in doubt about what specimens to collect, timing of specimen collection, or where or how to transport specimens, call the state health department and the state laboratory.
Table 3. Specimen collection for laboratory testing for VPDs
| Disease | Test name | Specimens to take | Timing for specimen collection | Transport requirements | Collection requirements |
|---|---|---|---|---|---|
| Diphtheria |
Culture Note: ALERT lab that diphtheria is suspected, so that tellurite-containing media will be used. |
Swab of nose, throat, membrane | ASAP, when diphtheria is suspected | < 24 hrs: Amies’ or similar transport medium ≥24 hrs: silica gel sachets |
State health departments may call CDC diphtheria lab at 404-639-1231 or 404-639-1239. |
| Diphtheria |
PCR Note: ALERT lab that diphtheria is suspected, so that specific PCR assay will be used. |
Swabs (as above), pieces of membrane, biopsy tissue | Take these specimens at same time as those for culture. | Silica gel sachet; or a sterile dry container at 4°C | State health departments may call CDC diphtheria lab at 404-639-1231 or 404-639-1239. |
| Diphtheria | Toxigenicity testing (Elek test) | Isolate from culture (above) | After C. diphtheriae has been isolated | Transport medium such as Amies medium, or silica gel sachets | State health departments may call CDC diphtheria lab at 404-639-1231 or 404-639-1239. |
| Diphtheria |
Serology (antibodies to diphtheria toxin) Note: Collect paired sera, taken 2-3 weeks apart. |
Serum | Before administration of antitoxin | Frozen (-20°C) | |
| Haemophilus influenzae type b |
Culture Note: Request that lab conduct serotyping on any H. influenzae isolate from any normally sterile site. |
Blood | ASAP | Blood culture bottles w/broth or lysis-centrifugation tube | Collect 3 separate samples in a 24-hr period. |
| Haemophilus influenzae type b |
Culture Note: Request that lab conduct serotyping on any H. influenzae isolate from any normally sterile site. |
CSF | ASAP | Sterile, screw-capped tube | |
| Haemophilus influenzae type b | Culture | Other normally sterile site | ASAP | ||
| Haemophilus influenzae type b | Serotyping | Isolate from culture (above) | Highest priority are isolates from persons <15 years. | ||
| Haemophilus influenzae type b | Antigen detection | Any normally sterile site | ASAP | ||
| Hepatitis A | IgM anti-HAV | Serum | ASAP after symptom onset (detectable up to 6 months) | All sera to be tested for serologic markers of HAV and HBV infection can be kept at ambient temperatures, refrigerated, or frozen for short term (<48 hours). For longer than 48 hours storage, sera should be frozen or refrigerated. | Non-hemolyzed |
| Hepatitis A |
Total anti-HAV Note: Measures both IgM and IgG. |
Serum | No time limit | Non-hemolyzed | |
| Hepatitis B | IgM anti-HBc | Serum | ASAP after symptom onset (Detectable up to 6 months) | Non-hemolyzed | |
| Hepatitis B |
HBsAg Note: HBsAg-positive results should be confirmed by HBsAg neutralization assay as specified in the package insert for each assay. |
Serum | Non-hemolyzed | ||
| Hepatitis B | Anti-HBs | Serum | 1-2 months after vaccination | Non-hemolyzed | |
| Influenza | Culture/viral isolation | Nasal wash, nasopharyngeal aspirates, nasal/throat swabs, transtracheal aspirate, bronchoalveolar lavage | Within 72 hours of onset of illness | Transport specimens at 4°C if tests are to be performed within 72 hours; otherwise, freeze at -70°C until tests can be performed. | |
| Influenza | Antigen detection and RT-PCR | Nasal wash, nasopharyngeal aspirate, nasal/throat swabs, gargling fluid, transtracheal aspirates, bronchoalveolar lavage | Within 72 hours of onset of illness | Transport specimens at 4°C if tests are to be performed within 72 hours; otherwise, freeze at -70°C until tests can be performed. | Note: Save an aliquot of the clinical sample for confirmation and isolation. Viral isolates may be further characterized by WHO/CDC. |
| Influenza | Serology | Paired sera | Acute: within 1 week of onset Convalescent: 2-3 weeks after acute |
Store at 4°C or frozen | Note: Fourfold rise is a positive result. Consider vaccination history. |
| Measles | Culture/PCR | Nasopharyngeal aspirates, throat swabs, urine, heparinized blood | Collect at same time as samples for serology (best within 3 days of rash onset) | Note: PCR for molecular typing. Do not collect if after 10 days from rash onset. | |
| Measles | IgM antibody | Serum | ASAP, and repeat 72 hours after onset if first negative | Note: IgM is detectable for at least 30 days after rash onset. | |
| Measles | IgG antibody | Paired sera | Acute: ASAP after rash onset (7 days at the latest) Convalescent: 14-30 days after acute |
||
| Meningococcal disease | Culture* | Blood | ASAP | TI medium is preferred. Blood culture bottles w/broth or lysis-centrifugation tube |
Note: Request that lab conduct serogrouping on any N. meningitidis isolate from any normally sterile site. |
| Meningococcal disease | Culture* | CSF | ASAP | TI medium is preferred. Sterile, screw-capped tube | Note: Request that lab conduct serogrouping on any N. meningitidis isolate from any normally sterile site. |
| Meningococcal disease | Culture* | Other normally sterile site | ASAP | TI medium is preferred. | |
| Meningococcal disease | Serogrouping | Isolate from culture (above) | Slant, frozen, lyophilized or silica gel pack. | ||
| Meningococcal disease | PCR | Any normally sterile site | ASAP | Sent frozen on blue ice packs. | |
| Mumps | Culture | Buccal /parotid swabs, CSF | Massage the salivary/parotid gland area for 30 seconds prior to swab collection. | ||
| Mumps | IgM antibody | Serum | ASAP; antibodies peak about a week after onset | ||
| Mumps | IgG antibody | Paired sera | Acute: within several days of onset Convalescent: 2 weeks after acute |
||
| Pertussis |
Culture Note: Inoculate selective primary isolation media such as charcoal horse blood agar or Bordet-Gengou as soon as possible. |
Posterior nasopharyngeal swab or aspirate | Within the first 2 weeks of cough onset | Swabs: half-strength charcoal horse blood agar at 4°C Aspirates: in catheter trap at 4°C |
Use Dacron or calcium alginate (not cotton) swabs with flexible shaft or aspiration by catheter attached to catheter trap. |
| Pertussis |
PCR Note: PCR should be validated with culture when possible. |
Nasopharyngeal swab or aspirate | Within the first 2 weeks of cough onset | Short term at 4°C; long term -20°C or below | Use Dacron (not calcium alginate or cotton) swabs with flexible shaft or aspiration by catheter attached to catheter trap. |
| Pertussis | Serology | Acute and convalescent sera | Acute: within the first 2 weeks of cough onset Convalescent: 3-9 weeks after acute |
-20°C | Note: Results are presumptive and should be validated with culture. Serologic results are currently not accepted as laboratory confirmation for purposes of national surveillance. |
| Pneumococcal disease | Culture | Normally sterile site | As soon as possible after onset of clinical illness but before administration of antibiotics | Blood culture bottles w/broth or lysis-centrifugation tube or, if from another sterile site, a sterile, screw-capped tube | Collect 2 separate blood samples in a 24-hr period. Most other sterile specimens (e.g., CSF) are collected only once. |
| Pneumococcal disease | PCR | Normally sterile site | ASAP, soon after administration of antibiotics is a viable option. | Send specimen frozen on blue ice packs | PCR |
| Pneumococcal disease | PCR deduction of serotype | Culture-negative sterile site specimen | Specimen frozen immediately | PCR deduction of serotype | |
| Pneumococcal disease | Susceptibility testing | Pure culture | Slant, frozen, or silica packet | Susceptibility testing | |
| Pneumococcal disease | Serotyping | Pure culture | Slant, frozen, or silica packet | Serotyping | |
| Poliomyelitis | Culture | Stool, pharyngeal swab, CSF | Acute | Sterile, screw-capped container |
No carrier for stool; saline buffer for swabs Note: Maintain frozen or transport rapidly to lab; avoid desiccation of swab specimens. |
| Poliomyelitis | Intratypic differentiation | Isolate from culture (above) | Note: Maintain frozen or transport rapidly to lab; avoid desiccation of swab specimens. | ||
| Poliomyelitis | Serology | Paired sera | Acute: ASAP Convalescent: 3 weeks after acute |
||
| Rotavirus gastroenteritis | EIA, PCR genotyping | Stool, sera if stool not available | First to fourth day of illness optimal (stool); third to seventh day (serum) | Sterile, screw-capped container |
Bulk stool, whole serum Note: Keep frozen or transport rapidly to lab; avoid multiple freeze-thaw cycles. |
| Rotavirus gastroenteritis | Culture, RNA electrophoresis, EM | Stool | First to fourth day of illness optimal | Sterile, screw-capped container |
Bulk stool, whole serum Note: Keep frozen or transport rapidly to lab; avoid multiple freeze-thaw cycles. |
| Rotavirus gastroenteritis | Serology | Paired sera | Acute: ASAP Convalescent: 3 weeks after acute |
Sterile, screw-capped container | Whole serum |
| Rotavirus-associated seizures | PCR | CSF | ASAP after symptoms begin | Sterile, screw-capped container |
No carrier Note: Keep frozen or transport rapidly to lab; avoid multiple freeze-thaw cycles. |
| Rubella | IgM antibody | Serum | Within 7-10 days of onset | ||
| Rubella | IgG antibody | Paired sera | Acute: within 7-10 days of onset of illness Convalescent: 2-3 weeks after acute |
||
| Rubella | Culture/PCR | Nasopharyngeal swab/wash, throat, urine. | Within 4 days of onset of rash | Viral transport media | Note: Maintain frozen (except urine) or transport rapidly to lab; avoid desiccation of swab specimens. |
| Congenital rubella syndrome (CRS) | IgM antibody | Serum | As soon as possible, within 6 months of birth | ||
| Congenital rubella syndrome (CRS) | IgG antibody | Paired sera | Note: Confirmation is by documenting persistence of serum IgG titer beyond the time expected from passive transfer of maternal IgG antibody. | ||
| Congenital rubella syndrome (CRS) | Culture/PCR | Nasopharyngeal swab/wash, urine, blood, cataracts | As soon as possible; every 1-3 months until cultures are repeatedly negative | Viral transport media | Note: Maintain frozen (except urine) or transport rapidly to lab; avoid desiccation of swab specimens. |
| Varicella | Serology | Serum | Immune status: collect anytime except during acute illness Paired serologic diagnosis: acute within 7-10 days of onset; convalescent 2-3 weeks after acute |
Single IgG assay is useful to assess immune status. Paired serum used to identify recent infection, but is not the method of choice when rapid diagnosis needed. |
|
| Varicella | Direct immuno-fluorescent antibody (DFA) | Scraping/swab from base of vesicle | Acute illness 2-3 days after rash onset and fresh vesicles | Note: Used for rapid diagnosis. | |
| Varicella | Culture | Fluid from vesicles, nasal or throat swabs, serum, spinal fluid, urine, bronchial tree washing or inflamed joints | Acute illness 2-3 days after rash onset and fresh vesicles | Note: Definitive diagnosis, but not useful for rapid diagnosis. | |
| Varicella | Viral typing/strain identification | Viral isolate (from culture) | Within 2-3 days of rash onset | Storage more than a few hours must be kept on dry ice or frozen at -70°C or below | Note: Merck and Co., Inc., offers a free viral identification service using PCR analysis (1-800-672-6372). |
* Neisseria meningitidis culture cannot be performed on specimens sent to CDC, but CDC is available to provide advice and answer questions on culture methods.
A. Diphtheria (see Chapter 1)
Diagnostic tests used to confirm infection include isolation of Corynebacterium diphtheriae on culture and toxigenicity testing. Although no other tests for diagnosing diphtheria are commercially available, CDC can perform a polymerase chain reaction (PCR) test on clinical specimens to confirm infection with a potentially toxigenic strain. PCR can detect nonviable C. diphtheriae organisms from specimens taken after antibiotic therapy has been initiated.
Although PCR for the diphtheria toxin gene and its regulatory element, as performed by the CDC Pertussis and Diphtheria Laboratory, provides supportive evidence for the diagnosis, data are not yet sufficient for PCR to be accepted as a criterion for laboratory confirmation. At present, a case that is PCR positive without the isolation of the organism or histopathologic diagnosis or without epidemiologic linkage to a laboratory-confirmed case should be classified as a probable case.
Isolation of C. diphtheriae by culture
Isolation of C. diphtheriae by bacteriological culture is essential for confirming diphtheria. The following should be considered:
- A clinical specimen for culture should be obtained as soon as possible when diphtheria (involving any site) is suspected, even if treatment with antibiotics has already begun.
- Specimens should be taken from the nose and throat, and from the diphtheritic membrane.
If possible, swabs also should be taken from beneath the membrane. - The laboratory should be alerted to the suspicion of diphtheria because isolation of C. diphtheriae requires special culture media containing tellurite.
- Isolation of C. diphtheriae from close contacts may confirm the diagnosis of the case, even
if the patient’s culture is negative.
All suspected cases and their close contacts should supply specimens from the nose and throat (i.e., both a nasopharyngeal and a pharyngeal swab) for culture.
Biotype testing
After C. diphtheriae has been isolated, the biotype (substrain) should be determined. The four biotypes are intermedius, belfanti, mitis, and gravis.
Toxigenicity testing
In addition to determining biotype, toxigenicity testing using the Elek test should be performed to determine if the C. diphtheriae isolate produces toxin. These tests are not readily available in many clinical microbiology laboratories; isolates should be sent to a reference laboratory proficient in performing the tests.
Polymerase chain reaction testing
Additional clinical specimens for PCR testing at CDC should be collected at the time specimens are collected for culture. Because isolation of C. diphtheriae is not always possible (many patients have already received several days of antibiotics by the time a diphtheria diagnosis is considered), PCR can provide additional supportive evidence for the diagnosis of diphtheria. The PCR assay allows for detection of the regulatory gene for toxin production (dtxR) and the diphtheria toxin gene (tox).[6] Clinical specimens (swabs, pieces of membrane, biopsy tissue) can be transported to CDC with cold packs in a sterile empty container or in silica gel sachets. For detailed information on specimen collection and shipping and to arrange for PCR testing, the state health department may contact the CDC Pertussis and Diphtheria Laboratory at 404-639-1231 or 404-639-1239.
Serologic testing
Measurement of the patient’s serum antibodies to diphtheria toxin before administration of antitoxin may help in assessing the probability of the diagnosis of diphtheria. The state health department or CDC can provide information on laboratories that offer this test (few laboratories have the capability to accurately test antibody levels). If antibody levels are low, diphtheria cannot be accurately ruled out, but if levels are high, C. diphtheriae is less likely to produce serious illness.
Submission of C. diphtheriae isolates
All isolates of C. diphtheriae from any body site (respiratory or cutaneous), whether toxigenic or nontoxigenic, should be sent to the CDC Pertussis and Diphtheria Laboratory for reference testing. Clinical specimens from patients with suspected diphtheria to whom diphtheria antitoxin has been released for treatment should also be sent to the CDC Pertussis and Diphtheria Laboratory for culture and PCR testing. To arrange for shipping of specimens, contact your state health department.
B. Haemophilus influenzae type b (Hib) invasive disease (see Chapter 2)
Culture
Confirming a case of Hib disease requires culturing and isolating the bacterium from a normally sterile body site. Normally sterile site specimens include cerebrospinal fluid (CSF), blood, joint fluid, pleural effusion, pericardial effusion, peritoneal fluid, subcutaneous tissue fluid, placenta, and amniotic fluid. Most hospital and commercial microbiologic laboratories have the ability to isolate H. influenzae (Hi) from cultured specimens. All Hi isolates should also be tested for antimicrobial susceptibility according to guidelines in M02-A11 Performance Standards for Antimicrobial Disk Susceptibility Tests (January 2012) from the Clinical Laboratory Standards Institute.[7]
Serotype testing (serotyping)
Serotyping distinguishes encapsulated strains, including Hib, from unencapsulated strains, which cannot be typed. The six encapsulated types (designated a-f) have distinct capsular polysaccharides that can be differentiated by slide agglutination with type-specific antisera.
To monitor the occurrence of invasive Hib disease, microbiology laboratories should perform serotype testing of all H. influenzae isolates,[8, 9] particularly those obtained from children younger than 5 years of age. To monitor the disease burden and long-term vaccine effectiveness, Hi isolates from children aged 5-14 years should also be serotyped and reported. Even though Hib disease has declined, laboratories should continue routine serotyping. Contact your state health department if serotyping is not available at your laboratory. State health departments with questions about serotyping should contact the CDC Meningitis and Vaccine Preventable Disease Branch laboratory at 404-639-3158.
Antigen detection
Because the type b capsular antigen can be detected in body fluids, including urine, blood, and CSF of patients, clinicians often request a rapid antigen detection test for diagnosis of Hib disease. Antigen detection may be used as an adjunct to culture, particularly in the diagnosis of patients who have received antimicrobial agents before specimens are obtained for culture. Methods for antigen detection include latex agglutination (LA) and counterimmunoelectrophoresis. LA is a rapid and sensitive method used to detect Hib capsular polysaccharide antigen in CSF, serum, urine, pleural fluid, or joint fluid. Counterimmunoelectrophoresis is more specific but less sensitive than LA, but takes longer and is more difficult to perform.
If the Hib antigen is detected in CSF but a positive result is not obtained from culture or sterile site, the patient should be considered as having a probable case of Hib disease and reported as such. Because antigen detection tests can be positive in urine and serum of persons without invasive Hib disease, persons who are identified exclusively by positive antigen tests in urine or serum should not be reported as cases. PCR assays for Hib in clinical specimens are available for research purposes only.[10-12] Isolation of the bacterium is needed to confirm Hi invasive disease, determine the serotype, and test for antimicrobial susceptibility.
Subtyping
Although not widely available, subtyping the Hib bacterium by pulsed-field gel electrophoresis (PFGE),[13, 14] multilocus sequence typing (MLST), and 16S rRNA gene sequence typing can be performed for epidemiologic purposes. Some subtyping methods, such as outer membrane proteins, lipopolysaccharides, or enzyme electrophoresis, are no longer recommended or performed because they were unreliable or too labor intensive. The state health department may direct questions about subtyping to the CDC Meningitis and Vaccine Preventable Disease Branch laboratory at 404-639-3158.
C. Hepatitis A (see Chapter 3)
Diagnostic tests used to confirm hepatitis A virus infection include serologic testing, and occasionally, PCR-based assays to amplify and sequence viral genomes.
Serologic testing
The diagnosis of acute hepatitis due to hepatitis A virus (HAV) is confirmed during the acute or early convalescent phase of infection by the presence of IgM anti-HAV in serum.
Serum for IgM anti-HAV testing should be obtained as soon as possible after onset of symptoms because IgM anti-HAV generally disappears within 6 months after onset of symptoms.
IgG anti-HAV appears in the acute or convalescent phase of infection, remains for the lifetime of the person, and confers enduring protection against disease.
The antibody test for total anti-HAV measures both IgG anti-HAV and IgM anti-HAV. The presence of total anti-HAV and absence of IgM anti-HAV indicates immunity consistent with either past infection or vaccination. Commercial diagnostic tests are widely available for the detection of IgM and total (IgM and IgG) anti-HAV in serum.
CDC laboratory special studies
Occasionally, molecular virologic methods such as PCR-based assays are used to amplify and sequence viral genomes. These assays may be helpful to investigate common-source outbreaks of hepatitis A. Providers with questions about molecular virologic methods should consult with their state health department or the Division of Viral Hepatitis, Laboratory Branch, CDC.
D. Hepatitis B (see Chapter 4)
Diagnostic tests used to confirm hepatitis B virus (HBV) infection include serologic testing, genotyping and subtyping (in outbreak investigations), and occasionally PCR-based assays to amplify/quantify and determine the sequence of viral genomes.
Serologic testing
Several well-defined antigen-antibody systems are associated with HBV infection, including HBsAg and anti-HBs; hepatitis B core antigen (HBcAg) and antibody to HBcAg (anti-HBc); and hepatitis B e antigen (HBeAg) and antibody to HBeAg (anti-HBe). Serologic assays are commercially available for all of these except HBcAg because no free HBcAg circulates in blood.
The presence of HBsAg is indicative of ongoing HBV infection and potential infectiousness. In newly infected persons, HBsAg is present in serum 30-60 days after exposure to HBV. Anti-HBc develops in all HBV infections, appearing at onset of symptoms or liver test abnormalities in acute HBV infection, rising rapidly to high levels, and persisting for life. Acute or recently acquired infection can be distinguished by presence of the immunoglobulin M (IgM) class of anti-HBc, which persists for approximately 6 months. IgM anti-HBc may not be present in newly infected children younger than 2 years of age, especially if they acquired their infection through perinatal transmission.
In persons who recover from HBV infection, HBsAg is eliminated from the blood, usually in 2-3 months, and anti-HBs develops during convalescence. The presence of anti-HBs indicates immunity from HBV infection. After recovery from natural infection, most persons will be positive for both anti-HBs and anti-HBc, whereas only anti-HBs develops in persons who are successfully vaccinated against hepatitis B. Persons who do not recover from HBV infection and become chronically infected remain positive for HBsAg (and anti-HBc), although a small proportion (0.3% per year) of these persons may eventually clear HBsAg and develop anti-HBs.
In some cases, anti-HBc is the only serologic marker detected. Isolated anti-HBc can occur after HBV infection in persons who have recovered but whose anti-HBs levels have waned or in persons in whom anti-HBs failed to develop. Certain chronically infected persons may be positive for anti-HBc alone, with HBsAg levels that are below levels detectable by commercially available tests. Infants who are born to HBsAg-positive mothers and who do not become infected may also have detectable anti-HBc for up to 24 months after birth from passively transferred maternal antibody.
The diagnosis of acute hepatitis due to hepatitis B virus infection is serologically confirmed by a positive test for IgM antibody to hepatitis B core antigen (anti-HBc). If testing for IgM antiHBc is not available, the diagnosis of acute hepatitis B can also be confirmed by a positive test for hepatitis B surface antigen (HBsAg) with a negative test for hepatitis A antibody (anti-HAV) (Table 4). Confirmation of HBsAg-positive results by HBsAg neutralization assay should be done as needed according to the manufacturer’s instructions in the package insert. In addition to acute HBV infection, both perinatal HBV infection and chronic HBV infection are reportable vaccine-preventable conditions. Chronic infection with HBV is confirmed by a positive test for HBsAg accompanied by a negative test for IgM anti-HBc or by two positive HBsAg test results that are at least 6 months apart. A diagnosis of perinatal HBV infection is confirmed by a positive test for HBsAg in an infant aged 1-24 months born in the United States or in U.S. territories to an HBsAg-positive mother.
Table 4. Interpretation of hepatitis B serologic tests.
| Serologic Marker: HBsAg* | Serologic Marker: Total Anti-HBc † | Serologic Marker: IgM Anti-HBc § | Serologic Marker: Anti-HBs ¶ | Interpretation |
|---|---|---|---|---|
| - | - | - | - | Susceptible, never infected |
| + | - | - | - | Acute infection, early incubation** |
| + | + | + | - | Acute infection |
| - | + | + | - | Acute resolving infection |
| - | + | - | + | Past infection, recovered and immune |
| + | + | - | - | Chronic infection |
| - | + | - | - | False positive (i.e., susceptible), past infection, or ‘low level’ chronic infection |
| - | - | - | + | Immune if titer is >10 mIU/ml |
* Hepatitis B surface antigen
† Antibody to hepatitis B core antigen
§ Immunoglobulin M
¶ Antibody to hepatitis B surface antigen
** Transient HBsAg positivity (lasting less than 18 days) might be detected in some patients during vaccination.
Genotyping and subtyping
Genotyping and subtyping of HBsAg has occasionally been used to investigate outbreaks of hepatitis B, but this procedure is not routinely available in commercial laboratories.
Molecular analysis
Molecular virologic methods such as PCR-based assays are available from CDC and commercial laboratories for detection and sequencing of HBV DNA. Although results for HBV DNA are not currently included in the definition for acute hepatitis B, they are included for the chronic HBV definition. Testing for HBV DNA is most commonly used for the purpose of evaluating a patient with diagnosed HBV infection who is receiving or being considered for treatment; these tests are not typically used for the initial diagnosis of infection.
PCR-based methods for amplifying and sequencing the HBV genome, done in conjunction with epidemiologic studies, may be helpful for investigating common-source outbreaks of hepatitis B infection. In addition, these assays are essential for detecting the emergence of vaccine-resistant strains. For example, detection of HBV variants or “escape mutants” among vaccinated infants of HBsAg-positive women is important to determine their potential role in vaccine failures.[15] Healthcare professionals with questions about molecular virologic methods or those who identify HBsAg-positive events among vaccinated persons should consult with their state health department or the Epidemiology Branch, Division of Viral Hepatitis, CDC, 404-718-8500.
E. Influenza (see Chapter 6)
Methods available for the diagnosis of influenza include virus isolation (standard methods and rapid culture assays), molecular detection (reverse transcriptase-polymerase chain reaction [RT-PCR]), detection of viral antigens (enzyme immunoassays [EIA], immunofluorescent antibody [IFA], and commercially available rapid diagnostic kits), and less frequently, electron microscopy, and serologic testing.
Virus isolation
Virus isolation is the gold standard for influenza diagnosis. The following guidelines should be considered:
- Appropriate samples include nasal washes, nasopharyngeal aspirates, nasal and throat swabs, transtracheal aspirates, and bronchoalveolar lavage.
- Samples should be taken within 72 hours of onset of illness to maximize the probability of isolating virus.
- Rapid culture assays that use immunologic methods to detect viral antigens in cell culture are available. These assays can provide results in 18-40 hours, compared with an average of 4.5 days to obtain positive results from standard culture.
Molecular testing methods
RT-PCR, including real-time RT-PCR, can be used to detect the presence of influenza
virus in a clinical specimen or to characterize an influenza virus grown in tissue culture or embryonated eggs.
RT-PCR testing can be performed under biosafety level 2 conditions, even for viruses such as avian influenza A(H5N1), which require biosafety level 3 with enhancements for viral culture.
Antigen detection assays
Several methods exist for the diagnosis of influenza infection directly from clinical material:
- Cells from the clinical sample can be stained using an immunofluorescent antibody to look for the presence of viral antigen. Nasal washes, nasopharyngeal aspirates, nasal and throat swabs, gargling fluid, transtracheal aspirates, and bronchoalveolar lavage are suitable clinical specimens.
- Commercially available kits to test for the presence of viral antigens fall into three groups: the first detects only influenza type A viruses, while the second detects both influenza type A and B viruses but does not differentiate between virus types; and the third detects both influenza type A and B viruses and distinguishes between the two. Results of these rapid antigen detection tests can be available in less than 1 hour.
- Other less frequently used methods include immunostaining and visualization of viral antigens by electron microscopy.
- When direct antigen detection methods are used for the diagnosis of influenza, it is important to collect and reserve an aliquot of the clinical sample for possible further testing. The medium used to store the specimen for some rapid testing methods is inappropriate for viral culture; in this case, it is necessary to collect two separate samples. These additional or reserved samples may be used to confirm direct test results by culture and to subtype influenza A isolates.
Serologic testing
Serologic diagnosis of influenza infection requires paired serum specimens. The acute-phase sample should be collected within 1 week of the onset of illness, and the convalescent-phase sample should be collected approximately 2-3 weeks later.
Hemagglutination inhibition (HI) tests are the preferred method of serodiagnosis. A positive result is a fourfold or greater rise in titer between the acute- and convalescent-phase samples when tested at the same time. Serologic test results are usually available in 24 hours.
While serologic testing can be useful in certain situations where viral culture is not possible or in special studies, serologic diagnosis of influenza is not used for national surveillance because of the lack of standardized testing methods and interpretation.
F. Measles (see Chapter 7)
Serologic testing
Serologic testing for antibodies to measles is widely available. Generally, in a previously susceptible person exposed to either vaccine- or wild-type measles virus, the IgM response begins around the time of rash onset and can be detected for 1-2 months. The IgG response starts more slowly, at about 5-10 days after rash onset, but typically persists for a lifetime. The diagnosis of acute measles infection can be made by detecting IgM antibody to measles in a single serum specimen or by detecting a rise in the titer of IgG antibody in two serum specimens obtained approximately 2 weeks apart. Uninfected persons are IgM negative but will either be IgG negative or IgG positive, depending upon their previous infection or vaccination histories.
Recommendations for serologic testing for measles
- An enzyme immunoassay (EIA) test for IgM antibody to measles in a single serum specimen, obtained at the first contact with the suspected measles case-patient, is the recommended method for diagnosing acute measles.
- A single-specimen test for IgG is the most commonly used test for immunity to measles because IgG antibody is long-lasting.
- Testing for IgG along with IgM is recommended for suspected measles cases.
- Paired sera (acute and convalescent) may be tested for a rise in IgG antibody to measles to confirm acute measles infection.
- When a patient with suspected measles has been recently vaccinated (6-45 days prior to rash onset), neither IgM nor IgG antibody responses can distinguish measles disease from the response to vaccination. In this instance, a viral specimen should be obtained so CDC can attempt to distinguish between vaccine virus and wild-type virus (Table 5).
Table 5. Interpretation of measles enzyme immunoassay results*
| IgM Result | IgG Result | Previous infection history | Current infection | Comments |
|---|---|---|---|---|
| + | - or + | Not vaccinated, no prior history of measles | Recently received first dose of measles vaccine | Seroconversion. IgG response depends on timing of specimen collection. |
| + | - or + | Not vaccinated, no prior history of measles | Wild-type measles | Seroconversion. Classic clinical measles. IgG response depends on timing of specimen collection. |
| + | - or + | Previously vaccinated, primary vaccine failure | Recently received second dose of measles vaccine | Seroconversion. IgG response depends on timing of specimen collection. |
| - | + | Previously vaccinated, IgG+ | Recently received second dose of measles vaccine | IgG level may stay the same or may boost. |
| + | + | Previously vaccinated, IgG+ | Wild-type measles | May have few or no symptoms (e.g., no fever or rash). |
| + | + | Recently vaccinated | Exposed to wild-type measles | Cannot distinguish between vaccine or wild-type virus; evaluate on epidemiologic grounds.† |
| - | + | Distant history of natural measles | Vaccine | IgG level may stay the same or may boost. |
| + (at least in some patients) |
+ | Distant history of natural measles | Wild-type measles | May have few or no symptoms. |
* These results are those expected when using the capture IgM and indirect IgG enzyme immunoassays and may not apply to different assays due to different techniques and sensitivities/specificities.
† However, in this circumstance, IgM testing will be helpful. If negative, it could rule out wild-type measles infection.
Tests for IgM antibody. Although multiple possible methods exist for testing for IgM antibody, EIA is the most consistently accurate test and is therefore the recommended method. There are two formats for IgM tests. The first and most widely available is the indirect format, which requires a specific step to remove IgG antibodies. Problems with removal of IgG antibodies can lead to false-positive[16] or, less commonly, false-negative results.
The second format, IgM capture, does not require the removal of IgG antibodies. This is the preferred reference test for measles. One direct-capture IgM EIA is commercially available.
EIA tests for measles are often positive on the day of rash onset. However, in the first 72 hours after rash onset, up to 30% of tests for IgM may give false-negative results. Tests that are negative in the first 72 hours after rash onset should be repeated (Table 3); serum should be obtained for repeat testing 72 hours after rash onset. IgM is detectable for at least 28 days after rash onset and frequently longer.[17]
When a laboratory IgM test result is suspected of being false-positive (Table 3), additional tests may be performed. False-positive IgM results for measles may be due to the presence of rheumatoid factor in serum specimens. Serum specimens from patients with other rash illness, such as parvovirus B19, rubella, and roseola, have been observed to yield false-positive reactions in some IgM tests for measles. False-positive tests may be suspected when thorough surveillance reveals no source or spread of cases, when the case does not meet the clinical case definition, or when the IgG result is positive within 3 days of rash onset. In these situations, confirmatory tests may be done at the state public health laboratory or at CDC. IgM results by tests other than EIA can be validated with EIA. Indirect EIA tests may be validated with capture EIA.
Tests for IgG antibody. Because tests for IgG require two serum specimens and a confirmed diagnosis cannot be made until the second specimen is obtained, IgM tests are generally preferred. However, if the IgM tests remain inconclusive, a second (convalescent-phase) serum specimen, collected 14-30 days after the first (acute-phase) specimen, can be used to test for an increase in the IgG titer. These tests can be performed in the state laboratory or at CDC. A variety of tests for IgG antibodies to measles are available; these include EIA, hemagglutination inhibition, indirect fluorescent antibody tests, and plaque reduction neutralization. Complement fixation, although widely used in the past, is no longer recommended. The “gold standard” test for serologic evidence of recent measles virus infection is plaque reduction neutralization test of IgG in acute- and convalescent-phase paired sera.
Paired IgG testing for laboratory confirmation of measles requires the demonstration of a fourfold rise in titer of antibody against measles. The tests for IgG antibody should be conducted on both acute- and convalescent-phase specimens at the same time. The same type of test should be used on both specimens. The specific criteria for documenting an increase in titer depend on the test. EIA values are not titers, and increases in EIA values do not directly correspond to rises in titer.
Virus isolation
Isolation of measles virus in culture or detection of measles virus by RT-PCR in clinical specimens confirms the diagnosis of measles. However, since culture and RT-PCR can take weeks to perform, they are rarely useful in confirming an actual diagnosis of measles. Also, a negative culture or RT-PCR result does not rule out measles because the tests are greatly affected by the timing of specimen collection and the quality and handling of the clinical specimens. If positive, these tests can be useful adjuncts to diagnosing acute measles when serology results are inconclusive. If measles virus is cultured or detected by RT-PCR, the viral genotype can be used for molecular epidemiology and to distinguish between measles disease caused by a wild-type measles virus and a response to measles vaccination, caused by a vaccine strain.
Viral culture and RT-PCR are important for molecular epidemiologic surveillance to help determine:
- the origin of the virus,
- which viral strains are circulating in the United States, and
- whether these viral strains have become endemic in the United States.
Isolation of measles virus is technically difficult and is generally performed in research laboratories.
Specimens (urine, nasopharyngeal aspirates, heparinized blood, or throat swabs) from clinically suspected cases of measles obtained for virus culture should be shipped to the state public health laboratory or to CDC at the direction of the state health department as soon as measles is confirmed. Specimens should be properly stored while awaiting case confirmation (see Appendix 7[2 pages]). Clinical specimens for virus isolation should be collected at the same time as samples for serologic testing. Because virus is more likely to be isolated when the specimens are collected within 3 days of rash onset, collection of specimens for virus isolation should not be delayed until laboratory confirmation is obtained. Clinical specimens should ideally be obtained within 7 days of rash onset and should not be collected if more than 10 days have passed after rash onset.
G. Neisseria meningitiditis, Meningococcal disease (see Chapter 8)
Neisseria meningitidis is a gram-negative, encapsulated, aerobic diplococcus. Thirteen different meningococcal serologic groups have been defined, five of which (A, B, C, Y, and W135) cause the great majority of disease. The distinction between serogroups is based on the immunochemistry of the capsular polysaccharide, but more recently polymerase chain reaction (PCR) of capsule biosynthesis genes has been used for serogroup determination of isolates.[18] Serogroup A, C, Y, and W135 polysaccharides all elicit a serogroup-specific immune response, which allows for serogroup-specific vaccines. The serogroup B capsular polysaccharide is poorly immunogenic, thus making it challenging to develop a vaccine to protect against this serogroup. Vaccine development efforts for serogroup B are focusing on outer membrane proteins (OMPs) or other surface molecules rather than the capsular polysaccharide.[19]
Identification of N. meningitidis
The case definition for confirmed meningococcal disease requires isolation of N. meningitidis from a normally sterile site. Typically, the isolate comes from blood or cerebrospinal fluid (CSF), but it can also be from joint, pleural, or pericardial fluid. Aspirates or skin biopsies of purpura or petechiae can yield meningococci in cases of meningococcemia. The typical media used to grow the organism are chocolate agar or Mueller-Hinton medium in an atmosphere containing 5% carbon dioxide.[20] Gram staining for N. meningitidis is commonly used and continues to be a reliable and rapid method for presumptive identification. Intracellular gram-negative diplococci in CSF can be considered meningococci until proven otherwise.
In addition to bacteriology for definitive detection and identification, latex agglutination can be used for rapid detection of meningococcal capsular polysaccharides in CSF; however, false-negative or false-positive results can occur. Antigen agglutination tests on serum or urine samples are unreliable for the diagnosis of meningococcal disease.[21]
Real-time PCR detects DNA of meningococci in blood, CSF, or other clinical specimens. A major advantage of PCR is that it allows for detection of N. meningitidis from clinical samples in which the organism could not be detected by culture methods, such as when a patient has been treated with antibiotics before a clinical specimen is obtained for culture. Even when the organisms are nonviable following antimicrobial treatment, PCR can still detect N. meningitidis DNA.[18] Because of the severity of meningococcal disease, it is critical to treat the patient as soon as infection is suspected and not delay to obtain a culture or laboratory results.
Susceptibility testing
Routine antimicrobial susceptibility testing of meningococcal isolates is not recommended. N. meningitidis strains with decreased susceptibility to penicillin G have been identified sporadically from several regions of the United States, Europe and Africa.[22] Most of these isolates with reduced penicillin susceptibility remain moderately susceptible (minimum penicillin inhibitory concentration of between 0.12 µg/mL and 1.0 µg/mL). High-dose penicillin G remains an effective treatment against moderately susceptible meningococci. Surveillance of susceptibility patterns in populations should be conducted to monitor trends in N. meningitidis susceptibility.
Testing during outbreaks
Phenotypic and genotypic methods are used to investigate meningococcal diversity. Capsular polysaccharide (serogroup), porin protein PorB (serotype), and porin protein PorA (serosubtype) are all phenotypic characteristics that can be used to distinguish meningococci from one another.[19] Outbreaks of meningococcal disease are usually caused by the same or closely related strains.[23] Molecular genotyping techniques such as pulsed-field gel electrophoresis (PFGE), 16S rRNA gene sequencing, or multilocus sequence typing (MLST) are used for subtype characterization of an outbreak clone.[24, 25] This subtyping helps to better define the extent of the outbreak. It is crucial to have rapid and reliable results in determining the meningococcal serogroup in an outbreak because public health response will differ for vaccine-preventable or nonvaccine-preventable disease. Molecular genotyping provides important tools for understanding the overall epidemiology of meningococcal disease, but different methods may be more useful in certain settings. PFGE or 16S rRNA gene typing seem to be most useful for outbreak and short-time-period epidemiology, whereas MLST has become the “gold standard” for long-term, global tracing of meningococcal population changes.
H. Mumps (see Chapter 9)
Acute mumps infection can be confirmed by the presence of serum mumps IgM, a significant rise in IgG antibody titer in acute- and convalescent-phase serum specimens, positive mumps virus culture, or detection of virus by RT-PCR.
Serum should be collected as soon as possible after onset of parotitis for IgM testing or as the acute-phase specimen for determining seroconversion. The convalescent-phase specimen for IgG detection should be obtained about 2 weeks later. IgM antibodies are detectable within 5 days after onset of symptoms, reach a maximum level about a week after onset of symptoms, and remain elevated for several weeks or months.[26, 27] The timing of the IgM response to mumps infection in vaccinated persons is highly variable and may be delayed. Virus may be isolated from the buccal mucosa from 6 days before until 10 days after salivary enlargement. Urine is less likely than oral specimens to contain sufficient virus for culture or detection; therefore, buccal swabs are preferred.[28] However, maximal viral shedding occurs 1-3 days prior to onset and through day 5 following onset of symptoms. Virus may be cleared more rapidly from vaccinated persons who become infected, so early collection of viral samples is critical. Prior immunization against mumps or previous natural infection may be documented by the presence of serum IgG mumps-specific antibodies by EIA. The level of IgG, as measured by EIA, that provides immunity has not been established.
Serologic testing for IgM antibody
The serologic tests available for laboratory confirmation of mumps acute infections and immunity vary among laboratories. The state health department can provide guidance on available laboratory services and preferred tests.
Enzyme immunoassay. EIA is a highly specific test for diagnosing acute mumps infection At the direction of the state health department, healthcare providers and state and local health departments may send serum specimens from persons with suspected mumps cases to the CDC Measles, Mumps, Rubella & Herpes Virus Laboratory Branch for IgM detection by EIA.
Immunofluorescence assay (IFA). IFA assays have the advantage of being relatively inexpensive and simple. The reading of IFA IgM tests requires considerable skill and experience since nonspecific staining may cause false-positive readings.
Note: Commercially available IFA antibody assays and EIA kits for detection of mumps IgM are currently not FDA approved. Each laboratory must validate these tests independently.
Viral cultures
Mumps virus can be isolated from fluid collected from the parotid duct, other affected salivary gland ducts, throat, CSF and urine, although urine is unlikely to yield virus and therefore not recommended. Parotid duct swabs yield the best sample, particularly when the salivary gland area is massaged approximately 30 seconds prior to collection of the buccal/parotid duct fluid. An effort should be made to obtain the specimen as soon as possible after parotitis or meningitis onset. Because few laboratories perform mumps virus culture, it is rarely used for clinical diagnosis in uncomplicated cases. Successful isolation should always be confirmed by immunofluorescence with a mumps-specific monoclonal antibody or by molecular techniques. Molecular typing of virus isolates provides epidemiologically important information and is now recommended (see below).
Molecular typing
Molecular techniques such as RT-PCR can be used to detect mumps RNA for mumps confirmation in appropriately collected specimens. Molecular epidemiologic surveillance makes it possible to build a sequence database that will help track transmission pathways of mumps strains circulating in the United States. In addition, typing methods are available to distinguish wild-type mumps virus from vaccine virus. Specimens for molecular typing should ideally be obtained as soon as possible after the onset of parotitis, ideally from the day of onset to 3 days later (not more than 10 days after parotitis). Specific instructions for specimen collection and shipping may be obtained from CDC by contacting the Measles, Mumps, Rubella & Herpes Virus Branch at 404-639-1156/3512. Specimens for virus isolation and molecular typing should be sent to CDC as directed by the state health department.
I. Pertussis (see Chapter 10)
Culture
The preferred laboratory test for diagnosis of pertussis is isolation of Bordetella pertussis by bacterial culture.
Isolation of the B. pertussis bacterium is required to test for antimicrobial resistance and for molecular typing by PFGE. Although bacterial culture is specific for the diagnosis, it is relatively insensitive. Under optimal conditions 80% of suspected cases in outbreak investigations can be confirmed by culture; in most clinical situations isolation rates are much lower.[29] The timing of specimen collection can affect the isolation rate, as can inadequately collected specimens and concurrent use of effective antimicrobial agents. Because patients can remain culture positive even while taking effective antibiotics (e.g., when strains are resistant to the antibiotic), nasopharyngeal swab for culture should be obtained regardless of concurrent use of an antibiotic.
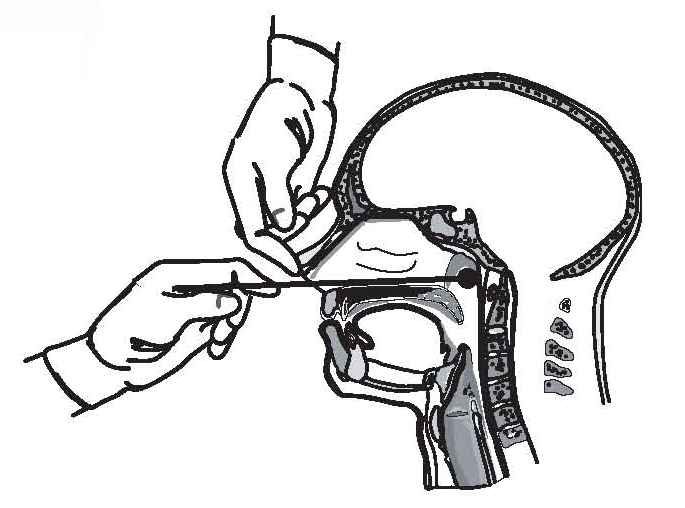
Fastidious growth requirements make B. pertussis difficult to isolate. Isolation of the organism using direct plating is most successful during the catarrhal stage (i.e., first 1-2 weeks of cough). All persons with suspected pertussis disease should have a nasopharyngeal aspirate or swab obtained from the posterior nasopharynx for culture. B. pertussis recovery rates from nasopharyngeal aspirates are similar to or higher than rates of recovery from nasopharyngeal swabs;[29-32] rates of recovery from throat and anterior nasal swabs are unacceptably low. Therefore, specimens should be obtained from the posterior nasopharynx (Figure 1), not the throat, by using Dacron® or calcium alginate swabs, not cotton. Specimens should be plated directly onto selective culture medium or placed in transport medium. Regan-Lowe agar or freshly prepared Bordet-Gengou medium generally is used for culture; half-strength Regan-Lowe can be used as the transport medium. Success in isolating the organism declines if the patient has received prior antibiotic therapy effective against susceptible B. pertussis (erythromycin or trimethoprim-sulfamethoxazole), if there is a delay in specimen collection beyond the first 2 weeks of illness, or if the patient has been vaccinated. A positive culture for B. pertussis confirms the diagnosis of pertussis. For this reason, access to a microbiology laboratory that is prepared to perform this service for no cost or for limited cost to the patient is a key component of pertussis surveillance.
Polymerase chain reaction
PCR testing of nasopharyngeal swabs or aspirates can be a rapid, sensitive, and specific method for diagnosing pertussis.[33] However, false-positive results may be obtained because of contamination in the laboratory or during specimen collection.[33, 34] PCR is currently available in some laboratories; the assay varies among laboratories and is not standardized. Direct comparison with culture is necessary for validation. Even if a laboratory has validated its PCR method, the result should be considered presumptive, and isolation of B. pertussis by culture should always be attempted to ensure that the disease is truly pertussis. B. pertussis isolates can then be evaluated for erythromycin susceptibility and by PFGE, which can help define the molecular epidemiology of strains circulating in the United States. Calcium alginate swabs are not acceptable for collecting specimens for PCR.
Serologic testing
Although serologic testing has proved useful in clinical studies, it is not yet standardized. Also, the lack of association between antibody levels and immunity to pertussis makes results of serologic testing difficult to interpret. For these reasons, serologic testing is not widely available. In Massachusetts, it is used for clinical diagnosis and reporting.[35] Elsewhere, with few exceptions, it is not known if serologic testing has been appropriately validated or standardized. Therefore, serologic testing should not be relied upon to confirm cases for the purpose of national reporting. Cases meeting the clinical case definition that are serologically positive, but not culture positive or PCR positive, should be reported as probable cases.
Direct fluorescent antibody testing
DFA testing of nasopharyngeal secretions may be useful as a screening test for pertussis. A positive DFA result may increase the probability that the patient has pertussis, but it has limited specificity (frequent false-positive results) and is not a confirmatory test. A monoclonal DFA test is available but the sensitivity and specificity are variable.
Elevated white blood cell count
An elevated white blood cell count with a lymphocytosis (i.e., increase in lymphocyte count) is usually present in cases of pertussis. The absolute lymphocyte count can reach 20,000/mm or higher. However, there may be no lymphocytosis in very young infants, vaccinated children, or adults with mild cases of pertussis. The white blood cell count is not a confirmation test.
Pulsed-field gel electrophoresis
Pulsed-field gel electrophoresis (PFGE) is a type of DNA fingerprinting. This technique has been useful tool for distinguishing epidemiologically related strains (e.g., strains from the same household or small community), while showing diversity within larger geographic areas such as cities, counties, and states.[36, 37]
Questions about performing PFGE on B. pertussis isolates, as well as questions about isolating B. pertussis, performing erythromycin susceptibility testing, and performing PCR can be directed to the Pertussis and Diphtheria Laboratory at CDC. Call Dr. M. Lucia Tondella at 404-639-1239, or Ms. Pam Cassiday at 404-639-1231. If needed, B. pertussis isolates can be sent to:
Attention: Pam Cassiday
DASH Unit 12
1600 Clifton Road NE
Atlanta, GA 30333
J. Pneumococcal infection (see Chapter 11)
Culture
Streptococcus pneumoniae is a gram-positive, lancet-shaped diplococcus that commonly inhabits the throat as normal flora. S. pneumoniae commonly causes lower and upper respiratory diseases, including pneumonia, meningitis and acute otitis media. Diagnosis of invasive pneumococcal infection is confirmed by culture and isolation of S. pneumoniae from a normally sterile body site (e.g., blood, CSF, pleural fluid, or peritoneal fluid). Alternatively, diagnosis can be confirmed from culture-negative specimens from normally sterile sites using real-time PCR.
Antibiotic resistance
The Clinical Laboratory Standards Institute (CLSI) recommends that clinical laboratories test all isolates of S. pneumoniae from CSF for resistance to penicillin, cefotaxime or ceftriaxone, meropenem, and vancomycin.[38] For organisms from other sources, laboratories should consider testing for resistance to erythromycin, penicillin, trimethoprim-sulfamethoxazole, clindamycin, cefepime, cefotaxime or ceftriaxone, a fluoroquinolone, meropenem, tetracycline, and vancomycin. Pneumococci resistant to vancomycin have never been described; a strain with a vancomycin minimum inhibitory concentration of 2 μg/ml or greater or zone diameter less than 17 mm should be submitted to a reference laboratory for confirmatory testing, and if resistant, should be reported to the state health department. Because pneumococci are fastidious organisms, some susceptibility testing methods used for other organisms are not appropriate for pneumococci; see the CLSI document for testing recommendations.[38]
Serotyping
Current pneumococcal vaccines are based upon capsular polysaccharides. There are currently 91 known capsular serotypes. Since only subsets of capsular serotypes are included in pneumococcal vaccines, serotyping allows the measurement of vaccine efficacy and can provide data for development of expanded-serotype vaccines.[39] CDC and its partners perform active, population-based surveillance for invasive pneumococcal serotypes in specific areas that represent about 30 million people in the United States. CDC does not provide serotyping outside of this surveillance except in specific situations, and this must first be cleared with Dr. Bernard Beall or a representative of the CDC Respiratory Diseases Branch Epidemiology section. Since typing sera are expensive and serotyping is technically difficult, detailed protocols are provided for variations of a simple PCR-based method for serotype deduction and in several publications.[40-43]
K. Poliomyelitis (see Chapter 12)
Virus isolation
The likelihood of poliovirus isolation is highest from stool specimens, intermediate from pharyngeal swabs, and very low from blood or spinal fluid. Isolation of poliovirus from stool specimens contributes to the diagnostic evaluation but does not constitute proof of a causal association between the isolated viruses and paralytic poliomyelitis.[44] Isolation of virus from CSF is diagnostic but is rarely accomplished. To increase the probability of poliovirus isolation, at least two stool specimens and two throat swabs should be obtained 24 hours apart from patients with suspected poliomyelitis as early in the course of the disease as possible (i.e., immediately after poliomyelitis is considered as a possible differential diagnosis), but ideally within the first 15 days after onset of paralytic disease. Specimens should be sent to the state or other reference laboratories for primary isolation. Laboratories should forward isolates to CDC for intratypic differentiation to determine whether the poliovirus isolate is wild or vaccine-derived.
Isolation of wild poliovirus constitutes a public health emergency, and appropriate control efforts must be immediately initiated (in consultation among healthcare providers, the state and local health departments, and CDC).
Serologic testing
Serology may be helpful in supporting or ruling out the diagnosis of paralytic poliomyelitis. An acute-phase serum specimen should be obtained as early in the course of disease as possible, and a convalescent-phase specimen should be obtained at least 3 weeks later. A fourfold rise in titer between the acute- and convalescent-phase specimens suggests poliovirus infection. Nondetectable antibody titers in both specimens may help rule out poliomyelitis but may be falsely negative in immunocompromised persons, who are also at highest risk for paralytic poliomyelitis. In addition, neutralizing antibodies appear early and may be at high levels by the time the patient is hospitalized, so that a fourfold rise may not be demonstrated. Vaccinated persons would also be expected to have measurable titers; therefore, vaccination history is important for interpretation of serologic tests. One of the limitations of serology is the inability to distinguish between antibody induced by vaccine-related poliovirus and antibody induced by wild virus. Serologic assays to detect anti-poliovirus antibodies are available in most commercial and state public health laboratories.
L. Rotavirus (see Chapter 13)
Laboratory testing is necessary to confirm group A rotavirus infection and to ensure reliable surveillance and clinical therapy. Because rotavirus is shed in such high concentrations in stool, fecal specimens are preferred for diagnosis of rotavirus. Methods available to diagnose rotavirus infection include detection of viral antigens (EIA, immunochromatography, electron microscopy, and immunostaining) and molecular detection by RT-PCR and nucleotide sequencing. [45] Serologic testing, although less commonly used, can detect a rise in serum IgG and IgA antibodies for recent infections.
Detection of viral antigens
The most widely available method of antigen detection in stool is EIA, which detects an antigen common to all group A rotaviruses.[45] Several inexpensive commercial EIA kits are available and provide rapid and highly sensitive results (90%-100%). Because EIA is rapid, inexpensive and highly sensitive, it is the most appropriate method for clinical diagnosis and surveillance.
Serotyping and subgrouping can be carried out using EIA methods. Monoclonal antibody-based EIA techniques have been invaluable in defining four globally common rotavirus serotypes (G1-G4) that represent more than 90% of the circulating strains and make up four of the five serotypes in the Rotateq® vaccine.[46] Two subgroups can also be differentiated by EIA techniques based on the reactivity of different monoclonal antibodies with the major capsid antigen that is common to all group A rotaviruses.
Another less frequently used method more appropriate for a research setting is visualization of viral particles by electron microscopy.
Molecular detection
Several molecular methods can be used to detect rotavirus infection in a clinical specimen and to characterize the virus, but these are used most commonly in research settings. Molecular methods for detection of viral RNA include RT-PCR, nucleotide sequencing, hybridization and silver staining.[45, 47]
- In recent years, multiplexed, semi-nested RT-PCR genotyping and nucleotide sequencing have become widely used to identify the most common and several uncommon rotavirus G and P genotypes. Hybridization can be used to confirm the results of RT-PCR genotyping.[45, 47]
- Nucleotide sequencing has been used extensively to identify uncommon strains and genetic variants that cannot be identified by RT-PCR genotyping and to confirm the results of genotyping methods.[45, 47]
- Nucleic acid hybridization is a less commonly used method to genotype rotaviruses.
- Electrophoresis and silver staining of viral RNA extracted from fecal specimens is a commonly used method for detection of rotavirus in research settings.
Virus isolation
Rotavirus can be isolated directly from fecal specimens by inoculation of cell cultures in the presence of trypsin-containing growth medium. This procedure is more appropriate for research laboratories.
Serologic testing
Routine diagnostic testing for rotavirus infection is primarily based on fecal specimen testing, although rotavirus antigen has been identified in serum samples of patients within 3-7 days of disease onset. Rotavirus diagnosis using serum specimens may prove especially valuable when fecal specimens are not available.[46] Serologic methods most commonly used to detect recent infections are EIA methods that detect a rise in serum IgG and IgA antibodies. In vaccine trials, the immunogenicity of rotavirus vaccines has been assessed by measuring rotavirus-specific IgG, IgA and neutralizing antibodies to vaccine strains.
M. Rubella (see Chapter 14)
Diagnostic tests used to confirm acute or recent rubella infection or congenital rubella syndrome (CRS) include serologic testing and virus isolation.
Serologic testing
Sera should be collected as early as possible (within 7-10 days) after onset of illness, and again at least 7-14 days (preferably 2-3 weeks) later. IgM antibodies may not be detectable before day 5 after rash onset. In case of a negative rubella IgM and IgG in specimens taken before day 5, serologic testing should be repeated. Virus may be isolated from 1 week before to 2 weeks after rash onset. However, maximum viral shedding occurs up to day 4 after rash onset.
False-positive serum rubella IgM tests have occurred in persons with parvovirus infections or positive heterophile test (indicating infectious mononucleosis) or with a positive rheumatoid factor (indicating rheumatologic disease).[48, 49] When a false-positive rubella IgM is suspected, a rheumatoid factor, parvovirus IgM, and heterophile test should be done to rule out a false-positive rubella IgM test result.
The serologic tests available for laboratory confirmation of rubella infections and immunity vary among laboratories. The following tests are widely available and may be used for screening for rubella immunity and/or laboratory confirmation of disease. The state health department can provide guidance on available laboratory services and preferred tests.
- Enzyme immunoassay. Most of the diagnostic testing done for rubella antibodies use some variation of the EIA, which is sensitive, widely available, and relatively easy to perform. EIA is the preferred testing method for IgM, using the capture technique; indirect assays are also acceptable.
- Hemagglutination inhibition (HI) test. HI once was the gold standard and most commonly used technique for confirmation of rubella infections. It allows for either screening or diagnosis (if paired acute- and convalescent-phase sera are tested). A fourfold rise or greater
in HI antibody titer in paired sera is diagnostic of recent infection. The test may be modified to detect rubella-specific IgM antibody, indicative of primary infection. - Latex agglutination (LA) test. The 15-minute LA test appears to be sensitive and specific for screening when performed by experienced laboratory personnel.
- Immunofluorescent antibody (IFA) assay. IFA is a rapid and sensitive assay. Commercial assays for both IgG and IgM are available in the United States. Care must be taken with the IgM assay to avoid false-positive results due to complexes with rheumatoid antibody.
Virus isolation
Rubella virus can be isolated from nasal, throat, urine, and cataract specimens from persons with rubella or CRS. The best results come from throat swabs. Efforts should be made to obtain clinical specimens for virus isolation from all case-patients (or from at least some patients in each outbreak) at the time of the initial investigation. Virus may be isolated from 1 week before to 2 weeks after rash onset. However, maximum viral shedding occurs up to day 4 after rash onset.
Molecular typing
Rubella virus isolates are very important for surveillance. Molecular epidemiologic surveillance provides important information on the origin of the virus, which virus strains are circulating in the United States, and whether these strains have become endemic in the United States.
In obtaining specimens for rubella molecular typing, collect throat swabs within 4 days of rash onset. Specimens for molecular typing from CRS patients should be collected as soon as possible after diagnosis. Appropriate specimens from CRS patients for molecular typing include throat/nasal swabs, urine, and cataracts from surgery. Specimens for virus isolation should be sent to CDC for molecular typing as directed by the state health department.
Reverse transcription polymerase chain reaction
In the United Kingdom, RT-PCR has been evaluated extensively for its usefulness in detection of rubella virus in clinical specimens.[50, 51] Clinical specimens obtained for virus isolation and sent to CDC are routinely screened by RT-PCR.
N. Congenital Rubella Syndrome (see Chapter 15)
Diagnostic tests used to confirm CRS include serologic assays and isolation of the virus. Laboratory confirmation can be obtained by any of the following methods:
- Demonstration of rubella-specific IgM antibodies in the infant’s cord blood or serum. In infants with CRS, IgM antibody persists for at least 6-12 months. In some instances, IgM may not be detected until at least 1 month of age; thus, infants with symptoms consistent with CRS who test negative shortly after birth should be retested at 1 month of age.[52]
- Documentation of persistence of serum rubella IgG titer beyond the time expected from passive transfer of maternal IgG antibody.
- Isolation of rubella virus. (Virus may be shed from the throat and urine for a year or longer, but best results come from specimens collected at or before 5 months of age.)
- Detection of rubella virus by RT-PCR.
O. Varicella (see Chapter 17)
Laboratory testing for varicella is not routinely required but is indicated to confirm the diagnosis in severe or unusual cases or to determine varicella susceptibility. Because varicella is the most common disease confused with smallpox, rapid laboratory confirmation of varicella zoster virus (VZV) diagnosis is required in cases of vesicular/pustular rash illness that fall into the category of “moderate risk” for smallpox according to the CDC algorithm. As disease continues to decline, laboratory confirmation will become standard practice. Diagnostic tests used to confirm recent varicella infection include virus isolation and identification, in addition to serologic tests.
Rapid varicella zoster virus identification
Rapid virus identification techniques are indicated for a case with severe or unusual disease to initiate specific antiviral therapy. The direct fluorescent antibody (DFA) test is the method of choice for rapid clinical diagnosis. This test is sensitive, specific, and widely available. Results are available within several hours. Specimens are best collected by unroofing a vesicle, preferably a fresh fluid-filled vesicle, and then rubbing the base of a skin lesion with a polyester swab. Crusts from lesions are also excellent specimens. Other specimen sources such as nasopharyngeal secretions, saliva, blood, urine, bronchial washings, and cerebrospinal fluid are considered less desirable sources than skin lesions since positive test results from such specimens are much less likely. Because viral proteins persist after cessation of viral replication, DFA may be positive when viral cultures are negative.
PCR
PCR is a powerful technique that permits the rapid amplification of specific sequences of viral DNA that would otherwise be present in clinical specimens at concentrations well below detectable limits. Carefully designed primers that target selected small stretches of viral DNA can be used to replicate small quantities of viral DNA extracted from clinical samples. If a PCR product of the expected size is produced, it is evidence that the virus was present in the lesion. This technique has been extended for VZV by amplifying pieces of varicella DNA that include a mutation in the base sequence that distinguishes the vaccine strain from wild-type varicella strains. Highly specific cutting enzymes (restriction endonucleases) can be selected that will cut the fragment from either wild-type strains or vaccine strain, but not both. This provides a convenient means for discriminating between them. More recently, it has been possible to apply these methods to real-time PCR machines that permit direct, single-step discrimination of vaccine strain from wild-type strains on the basis, for example, of the difference in temperature at which the strands from vaccine versus wild-type DNA fragments re-anneal on cooling. This type of approach has reduced the time required to identify a vaccine adverse event from 2 days to several hours.
Virus strain identification
Strain identification can distinguish wild-type VZV from the vaccine (Oka/Merck) strain using PCR and restriction fragment length polymorphism (RFLP) analysis. Such testing is important in situations when it is necessary to distinguish wild-type from vaccine-type virus in suspected vaccine adverse events. More recently, rapid real-time PCR methods using Light Cycler® or TaqMan® technology have made it possible to discriminate vaccine strain from wild-type VZV in a single tube assay requiring only a few hours. Postvaccination situations for which specimens should be tested include: 1) rash with more than 50 lesions occurring 7 or more days after vaccination, 2) suspected secondary transmission of the vaccine virus, 3) herpes zoster in a vaccinated person, or 4) any serious adverse event. The National VZV Laboratory at CDC has the capacity to distinguish wild-type VZV from Oka strain using both conventional and real-time PCR methods. Call the National VZV laboratory at 404-639-0066, 404-639-2192, or email dds1@cdc.gov or vzvlab@cdc.gov for details about collection and submission of specimens for testing.
Virus culture
The diagnosis of VZV infection may be confirmed by culture (isolation) of VZV. Although the virus is difficult to culture, virus isolation should be attempted in cases of severe disease, especially in immunocompromised persons, in order to confirm the diagnosis of varicella. Newer, more sensitive and rapid culture techniques can provide results within 2 to 3 days. Infectious VZV is usually recoverable from fluid from varicella lesions for 2 to 3 days and from zoster lesions for 7 days or longer. VZV may be cultured from other sites such as blood and CSF, especially in immunocompromised patients. Viable VZV cannot be recovered from crusted lesions.
Serologic testing
Serologic tests are available for IgG (acute and convalescent) and IgM antibodies to VZV for confirmation of disease. Testing using commercial kits for IgM antibody is not recommended since available methods lack sensitivity and specificity; false-positive IgM results are common in the presence of high IgG levels. The National VZV Laboratory at CDC has developed a reliable IgM capture assay. Call 404-639-0066, 404-639-2192, or email dds1@cdc.gov or kjr7@cdc.gov for details about collection and submission of specimens for testing.
Testing susceptibles
Single serologic IgG tests may be used to identify the immune status of persons whose history of varicella is negative or uncertain, and who may be candidates for varicella zoster immune globulin (VZIG) or vaccination. Paired acute- and convalescent-phase antibody tests are used in situations of mild or atypical presentation of disease when immediate therapy is not indicated and when, for clinical reasons, a confirmed diagnosis of the acute illness is important, e.g., a suspected second infection due to varicella. Recent evidence suggests that the latex agglutination method may result in false-positive tests that could mistakenly categorize a susceptible person as immune; less sensitive commercial ELISAs are recommended for the purpose of screening.[53] Routine testing for varicella immunity following vaccination is not recommended.
References
- CDC. Handbook of specimen collection and handling in microbiology. Atlanta: Centers for Disease Control, 1985.
- [CDC] Centers for Disease Control and Prevention. Measles lab manual from the World Health Organization (Isolation and identification of measles virus in cell culture) [updated 2009 AUG 31; accessed 2012 OCT 10]. Available at http://www.cdc.gov/measles/lab-tools/WHO-lab-manual.html.
- [CDC] Centers for Disease Control and Prevention. Laboratory protocols of rubella virus [updated 2008 JUL 28; accessed 2012 OCT 10]. Available at http://www.cdc.gov/rubella/lab/index.html.
- [CDC] Centers for Disease Control and Prevention. Division of Scientific Resources (DSR) [updated 2011 APR 8; accessed 2012 OCT 10]. Available at http://www.cdc.gov/ncezid/dsr/.
- [CDC] Centers for Disease Control and Prevention. Reference and disease surveillance.
- Mothershed EA, et al. Development of a real-time PCR assay for rapid detection of the diphtheria toxin gene. J Clin Microbiol 2002;40:4713-9.
- Clinical and Laboratory Standards Institute. M02-A11 Performance standards for antimicrobial disk susceptibility tests; Approved Standards-Eleventh Edition. Wayne, PA: Clinical and Laboratory Standards Institute, 2012.
- [CSTE] Council of State and Territorial Epidemiologists. Vaccine preventable disease surveillance and reporting. Position Statement ID-9 1999.
- CDC. Serotyping discrepancies in Haemophilus influenzae type b disease-United States, 1998-1999. MMWR 2002;51(32):706-7.
- Corless CE, et al. Simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in suspected cases of meningitis and septicemia using real-time PCR. J Clin Microbiol 2001;39:1553-8.
- Poppert S et al, Rapid diagnosis of bacterial meningitis by real-time PCR and fluorescence in situ hybridization. J Clin Microbiol 2005;43:3390-7.
- Tzanakaki G, et al. Simultaneous single-tube PCR assay for the detection of Neisseria meningitidis, Haemophilus influenzae type b and Streptococcus pneumoniae. Clin Microbiol Infect 2005;11:386-90.
- Fry AM, et al. Haemophilus influenzae type b disease among Amish children in Pennsylvania: reasons for persistent disease. Pediatrics 2001;108(4):E60.
- Lucher LA, et al. Reemergence, in southwestern Alaska, of invasive Haemophilus influenzae type b disease due to strains indistinguishable from those isolated from vaccinated children. J Infect Dis 2002;186:958-65.
- Thomas HC and Carman WF. Envelope and precore/core variants of hepatitis B virus. Gastroenterol Clin North Am 1994;23:499-514.
- Jenkerson SA, et al. False-positive rubeola IgM tests. N Engl J Med 1995;332:1103-1104.
- Helfand RF, et al. Diagnosis of measles with an IgM capture EIA: the optimal timing of specimen collection after rash onset. J Infect Dis 1997;175:195-9.
- Mothershed EA, et al. Use of real-time PCR to resolve slide agglutination discrepancies in serogroup identification of Neisseria meningitidis. J Clin Microbiol 2004;42:320-8.
- Raghunathan PL, et al. Opportunities for control of meningococcal disease in the United States. Annu Rev Med 2004;55:333-53.
- Granoff DM, et al. Meningococcal vaccines. In: Plotkin SA, Orenstein WA, Offit PA. Vaccines. 4th ed. Philadelphia, PA: Saunders; 2003:959-87.
- Rosenstein NE, et al. Meningococcal disease. N Engl J Med 2001;344:1378-88.
- American Academy of Pediatrics. 2006 Meningococcal infections. In: Pickering LK, et al, editors. Red book: 2006 Report of the Committee on Infectious Diseases. Elk Grove Village, IL: American Academy of Pediatrics; 2006. p. 452-60.
- CDC. Prevention and control of meningococcal disease. MMWR 2005;54(No. RR-7):1-17.
- Sacchi, CT, et al. Sequence diversity of Neisseria meningitidis 16S rRNA genes and use of 16S rRNA gene sequencing as a molecular subtyping tool. J Clin Microbiol
- Maiden, MC, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA 1998;95:3140-5.
- Ukkonen P, et al. Mumps-specific immunoglobulin M and G antibodies in natural mumps infection as measured by enzyme-linked immunosorbent assay. J Med Virol 1981;8:131-42.
- Benito RJ, et al. Persistence of specific IgM antibodies after natural mumps infection. J Infect Dis 1987;155:156-7.
- Tolpin MD, and Schauf V. Mumps virus 1984. Belsche RA, editor. Textbook of human virology. Littleton, MA: PSG Publishing Company, p. 311-32.
- Bejuk D, et al. Culture of Bordetella pertussis from three upper respiratory tract specimens. Pediatr Infect Dis J 1995;14:64-5.
- Halperin SA, et al. Evaluation of culture, immunofluorescence, and serology for the diagnosis of pertussis. J Clin Microbiol 1989;27:752-7.
- Hallander HO, et al. Comparison of nasopharyngeal aspirates with swabs for culture of Bordetella pertussis. J Clin Microbiol 1993;31:50-2.
- Hoppe JE and Weiss A. Recovery of Bordetella pertussis from four kinds of swabs. Eur J Clin Microbiol 1987;6:203-5.
- Meade BD and Bollen A. Recommendations for use of the polymerase chain reaction in the diagnosis of Bordetella pertussis infections. J Med Microbiol 1994;41:51-5.
- Lievano FA, et al. Issues associated with and recommendations for using PCR to detect outbreaks of pertussis. J Clin Microbiol. 2002;40:2801-5.
- Marchant CD, et al. Pertussis in Massachusetts, 1981-1991: incidence, serologic diagnosis, and vaccine effectiveness. J Infect Dis 1994;169:1297-1305.
- de Moissac YR, et al. Use of pulsed-field gel electrophoresis for epidemiological study of Bordetella pertussis in a whooping cough outbreak. J Clin Microbiol 1994;32:398-402.
- Bisgard KM, et al. Molecular epidemiology of Bordetella pertussis by pulsed-field gel electrophoresis profile: Cincinnati, 1989-1996. J Infect Dis 2001;183:1360-7.
- Clinical and Laboratory Standards Institute. M100-S22 Performance standards for antimicrobial susceptibility testing; Twenty-second informational supplement. Approved Standard. Wayne, PA: Clinical and Laboratory Standards Institute, 2012.
- Beall B, et al. Active Bacterial Core Surveillance Team. Pre- and postvaccination clonal compositions of invasive pneumococcal serotypes for isolates collected in the United States in 1999, 2001, and 2002. J Clin Microbiol 2006;44:999-1017.
- Pai R, et al. Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J Clin Microbiol 2006;44:124-31.
- Carvalho MG, et al. Evaluation and Improvement of real-time PCR detection assays to lytA, ply, and psaA genes for detection of pneumococcal DNA. J Clin Microbiol 2007. In press.
- Dias CA, et al. Sequential multiplex PCR for determining capsular serotypes of pneumococci recovered from Brazilian children. J Med Microbiol 2007;56:1185-8.
- Morais M, et al. Sequential multiplex PCR for identifying pneumococcal capsular serotypes from south-Saharan African clinical isolates. J Med Microbiol 2007;56:1181-4.
- Strebel PM, et al. Epidemiology of poliomyelitis in the United States one decade after the last reported case of indigenous wild virus-associated disease. Clin Infect Dis 1992;14:568-79.
- Gray JD and Desselberger U editors. 2000. Rotavirus, methods & protocols. (Methods in molecular medicine). New York: Humana Press, 2000.
- Estes MK and Kapikian AZ.. Rotaviruses. In: Knipe DM, et al, editors, Fields virology, 5th ed., vol. 2. Philadelphia: Lippincott Williams & Wilkins, 2007.p. 1917-74.
- Fischer TK and Gentsch JR. Rotavirus typing methods and algorithms. Rev Med Virol 2004;14:71-82.
- Kurtz JB and Anderson MJ. Cross-reactions in rubella and parvovirus specific IgM tests. Lancet 1985;2:1356.
- Morgan-Capner P. False positive tests for rubella-specific IgM. Pediatr Infect Dis J 1991;10:415-6.
- Bosma TJ, et al. PCR for detection of rubella virus RNA in clinical samples. J Clin Microbiol 1995;33:1075-9.
- Bosma TJ, et al. Use of PCR for prenatal and postnatal diagnosis of congenital rubella. J Clin Microbiol 1995;33:2881-7.
- CDC. Control and prevention of rubella: evaluation and management of suspected outbreaks, rubella in pregnant women, and surveillance for congenital rubella syndrome MMWR 2001;50(No. RR-12):1-23.
- Saiman L, et al. Persistence of immunity to varicella-zoster virus after vaccination of healthcare workers. Infect Control Hosp Epidemiol 2001;22:279-83.
- Page last reviewed: April 1, 2014
- Page last updated: November 9, 2015
- Content source: