Andersen–Tawil syndrome
| Andersen–Tawil syndrome | |
|---|---|
| Other names: Cardiodysrhythmic potassium-sensitive periodic paralysis, Andersen syndrome, long QT syndrome type 7, Andersen cardiodysrhythmic periodic paralysis[1] | |
 | |
| This condition affects the QT interval (in blue). | |
| Specialty | Cardiology, neurology, medical genetics |
| Symptoms | Abnormal heart rhythms, periodic paralysis, characteristic physical features[2] |
| Complications | Sudden cardiac death[3] |
| Usual onset | Present at birth; symptoms onset in childhood[3] |
| Duration | Lifelong |
| Types | Type 1 (KCNJ2 mutation), type 2 (genetic mutation not identified)[2] |
| Causes | Genetic (autosomal dominant)[2] |
| Diagnostic method | Based on symptoms, genetic testing[2] |
| Differential diagnosis | Thyrotoxic periodic paralysis, other causes of LQTS[1] |
| Treatment | Medication, implantable defibrillator[1] |
| Medication | Flecainide, potassium supplements, acetazolamide[1] |
| Frequency | ~1 in 1,000,000[4] |
Andersen–Tawil syndrome, also called long QT syndrome 7, is a genetic disorder.[2] Features include a long QT interval with a tendency for abnormal heart rhythms; episodes of muscle weakness known as periodic paralysis; abnormal facial features; and a short height.[3][2] Some degree of chronic weakness may also be present.[1] Symptoms generally begin in childhood; though some never develop symptoms.[3][1] Complications may include sudden cardiac death and scoliosis.[3][1]
In about 60% of cases it occurs due to a mutation in the KCNJ2 gene which encodes an ion channel that transports potassium.[3] Mutations in KCNJ5 may also result in the condition and in other cases the cause remains unknown.[5][2] Generally it is inherited from a person's parents in an autosomal dominant pattern.[3] Some cases occur due to a new mutation in early development.[3] Diagnosis is based on symptoms and may be supported by genetic testing.[2]
Management involves evaluation for certain conditions, treatment of specific symptoms when they occur, and efforts to prevent symptoms.[1] Periodic paralysis associated with a potassium of less than 3.0 mmol/L may be treated with 20 mEq potassium by mouth, which may be repeated.[1] Efforts to prevent attacks may include a carbonic anhydrase inhibitors such as acetazolamide or potassium supplements.[1] The abnormal heart rhythms may be treated with flecainide, though an implantable defibrillator may be required.[1] Medications that prolong the QT should be avoided.[1] Relatives should be checked for the condition.[1]
Andersen–Tawil syndrome is rare with about one person in every million affected.[4] Males and females are affected with similar frequency.[2] Early descriptions of the condition are believed to date from 1963 by Klein, followed by more detailed descriptions in 1971 by Ellen Andersen, and in 1994 by Rabi Tawil.[4]
Signs and symptoms
Andersen–Tawil Syndrome classically comprises three groups of features: abnormal electrical function of the heart, hypokalemic periodic paralysis, and characteristic physical features, although some of those affected will not exhibit all aspects of the condition.[6]
Andersen–Tawil syndrome affects the heart by prolonging the QT interval, a measure of how long it takes the heart to relax after each heart beat. This, as in other forms of long QT syndrome, can lead to abnormal heart rhythms such as ventricular ectopy or ventricular tachycardia causing palpitations.[6] The ventricular tachycardia seen in Andersen–Tawil syndrome often takes a form known as bidirectional ventricular tachycardia. The arrhythmias seen in association with the condition can cause sudden cardiac death, but the risk of this is lower than in other forms of long QT syndrome.[1] The physical abnormalities associated with Andersen–Tawil syndrome typically affect the head, face, limbs and spine. Abnormalities of the head and face include an unusually small lower jaw (micrognathia), low-set ears, widely spaced eyes (hypertelorism), a broad forehead and nasal root, a high arched or cleft palate, and a long narrow head (scaphocephaly).[4] Abnormalities of the limbs and spine include an abnormal curvature of the fingers, particularly the fifth finger (clinodactyly), fused fingers or toes (syndactyly), short stature, and a curved spine (scoliosis).[4]
The third key feature of Andersen–Tawil syndrome is intermittent muscle weakness. This can last from seconds to minutes, but in some cases may last for days at a time. Weakness often occurs at times when the levels of potassium in the blood are lower than normal (hypokalaemia), and is referred to as hypokalaemic periodic paralysis. This weakness can however occur at times when potassium levels are normal, triggered by other factors including exercise, cold, or even menstruation.[4]
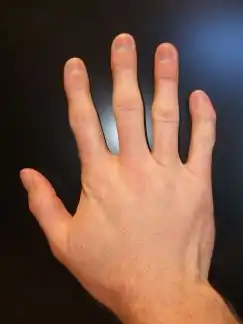 Clinodactyly – abnormal curvature of 5th finger towards 4th finger
Clinodactyly – abnormal curvature of 5th finger towards 4th finger Electrocardiogram showing bidirectional Ventricular Tachycardia in a 9-year-old female with Andersen–Tawil syndrome
Electrocardiogram showing bidirectional Ventricular Tachycardia in a 9-year-old female with Andersen–Tawil syndrome
Cause
Andersen–Tawil syndrome is a genetic disorder which in the majority of cases is caused by mutations in the KCNJ2 gene. The condition is often inherited from a parent in an autosomal dominant manner, but may occur due to a new genetic mutation in the affected person.[4]
Two types of Andersen–Tawil syndrome have been described, distinguished by the genetic abnormality that is detected. Type 1 Andersen–Tawil, accounting for about 60% of cases, is caused by mutations in the KCNJ2 gene.[7] In type 2 Andersen–Tawil, accounting for about 40% of cases, a KCNJ2 mutation is not identified. Mutations in a related gene encoding a similar potassium ion channel, KCNJ5, have been identified in some of those with type 2 Andersen–Tawil, but in many cases a genetic mutation is not found.[1]
The protein made by the KCNJ2 gene forms an ion channel that transports potassium ions into muscle cells. This specific channel (the inward rectifier potassium channel Kir2.1) carries a potassium current known as IK1 which is responsible for setting the resting membrane potential of muscle cells and is therefore critical for maintaining the normal functions of skeletal and cardiac muscle.[4] Pathogenic mutations in the KCNJ2 gene alter the usual structure and function of potassium channels or prevent the channels from being inserted correctly into the cell membrane. Many mutations prevent a molecule called PIP2 from binding to the channels and effectively regulating their activity. These changes disrupt the flow of potassium ions, leading to the periodic paralysis and abnormal heart rhythms characteristic of Andersen–Tawil syndrome.[7]
| Type | OMIM | Gene | Notes |
| Type 1 Andersen–Tawil syndrome | 170390 | KCNJ2 | Encodes inward rectifying potassium current Kir2.1 carrying the potassium current IK1.[1] |
| Type 2 Andersen–Tawil syndrome | 600734 | KCNJ5 | Also known as GIRK4, encodes G protein-sensitive inwardly rectifying potassium channels (Kir3.4) which carry the potassium current IK(ACh).[1] |
Mechanisms

Andersen–Tawil syndrome increases the risk of abnormal heart rhythms by disturbing the electrical signals that are used to coordinate individual heart cells. The genetic mutation disturbs an ion channel responsible for the flow of potassium, reducing the /K1 current. This prolongs of the cardiac action potential – the characteristic pattern of voltage changes across the cell membrane that occur with each heart beat, and depolarises the resting membrane potential of cardiac and skeletal muscle cells.[4]
Cardiac and skeletal muscle cells, when relaxed, have fewer positively charged ions on the inner side of their cell membrane than on the outer side, referred to as their membranes being polarised.[8] The main ion current responsible for maintaining this polarity is /K1, and a decrease in this current leads to less polarity at rest, or a depolarised resting membrane potential. When these cells contract, positively charged ions such as sodium and calcium enter the cell through ion channels, depolarising or reversing this polarity. After a contraction has taken place, the cell restores its polarity (or repolarises) by allowing positively charged ions such as potassium to leave the cell, restoring the membrane to its relaxed, polarised state.[8] The genetic mutation found in those with Andersen–Tawil decreases the flow of potassium, slowing the rate of repolarisation which can be seen in individual cardiac muscle cells as a longer action potential and on the surface ECG as a prolonged QT interval.[4]
The prolonged action potentials can lead to arrhythmias through several potential mechanisms. The frequent ventricular ectopy and bidirectional VT typical of Andersen–Tawil syndrome are initiated by a triggering beat in the form of an afterdepolarisation. Early afterdepolarisations, occurring before the cell has fully repolarised, arise due to reactivation of calcium and sodium channels that would normally be inactivated until the next heartbeat is due.[9] Under the right conditions, reactivation of these currents can cause further depolarisation of the cell, facilitated by the sodium-calcium exchanger.[9] Early afterdepolarisations may occur as single events, but may occur repeatedly leading to multiple rapid activations of the cell.[9] Delayed afterdepolarisations, occurring after repolarisation has completed, arise from the spontaneous release of calcium from the intracellular calcium store known as the sarcoplasmic reticulum. This calcium release then leaves the cell through the sodium calcium exchanger in exchange for sodium, generating a net inward current and depolarising the cell membrane.[9] If this transient inward current is large enough, a premature action potential is triggered.
The muscle weakness seen in those with Andersen–Tawil syndrome arises from the depolarisation of the resting membrane potential caused by a decrease in /K1.[4] The depolarised resting membrane potential means that sodium channels which are responsible for initiating action potentials are unable to fully recover from inactivation, leading to a less excitable membrane and less forceful muscle contraction.[4]
The mechanisms underlying the skeletal abnormalities seen in Andersen–Tawil syndrome have not been fully explained. Possibilities include impaired function of osteoclasts, cells which regulate bone growth, or disruption of the bone morphogenetic protein signalling cascade.[4]
Diagnosis

Andersen–Tawil syndrome is generally diagnosed based on symptoms, the findings on examination, and the results of an electrocardiogram.[4] Clinical diagnostic criteria have been proposed which suggest that a diagnosis can be made if two of the following four criteria are met: (1) periodic paralysis; (2) ventricular arrhythmias (frequent ventricular ectopic beats or ventricular tachycardia), a prolonged QT interval when corrected for rate, and/or a prominent U wave; (3) at least two of the following dysmorphic features: low-set ears, wide-set eyes, a small mandible, fifth-digit clinodactyly, and syndactyly; and (4) a family member with confirmed Andersen–Tawil syndrome.[4]
Genetic testing can be used to identify the specific mutation in an affected person, which if found can assist with screening family members.[4] Other investigations that may be helpful in making a diagnosis include ambulatory ECG monitoring to assess for arrhythmias, measurement of blood potassium levels at baseline and during periods of weakness, and measurement of thyroid function.[10]
Differential diagnosis
The differential diagnosis for a prolonged QT interval includes other forms of long QT syndrome such as Romano–Ward syndrome in which only the electrical activity of the heart is affected without involving any other organs; Jervell and Lange-Nielsen syndrome in which a prolonged QT interval is combined with congenital deafness; and Timothy syndrome in which a prolonged QT interval is combined with abnormalities in the structure of the heart, in addition to autism-spectrum disorder.[11] The frequent ventricular ectopy and bidirectional ventricular tachycardia seen in Andersen–Tawil syndrome can also occur in catecholaminergic polymorphic ventricular tachycardia.[6]
The intermittent weakness seen in Andersen–Tawil syndrome also occurs in other forms of periodic paralysis – hypokalaemic periodic paralysis, hyperkalaemic periodic paralysis, and paramyotonia congenita.[10]
Treatment
As a genetic condition, Andersen–Tawil syndrome cannot be cured. However, many of symptoms such as passing out due to abnormal heart rhythms and periodic paralysis can be successfully treated with medication or implantable devices. The rarity of the condition means that many of these treatments are based on consensus opinion as there are too few people to conduct adequately powered clinical trials.[4]
General measures
Medications should be avoided that further prolong the QT interval such as sotalol and amiodarone as these drugs can promote abnormal heart rhythms.[4] Lists of medications associated with prolongation of the QT interval can be found online.[12] Medications which reduce blood levels of potassium such as diuretics like furosemide and bendroflumethiazide should also be avoided as these can worsen the tendency to periodic paralysis and arrhythmias.[4] Conversely, potassium-containing supplements to increase blood potassium levels may be helpful.[4] Very strenuous or competitive sport should be discouraged as these may increase the risk of arrhythmias, although gentle exercise should be encouraged.[11]
Arrhythmias

The risk of arrhythmias may be reduced by taking beta blockers such as propranolol; however, the evidence for this is poor as of 2013.[4] Other antiarrhythmic drugs such as flecainide and verapamil may also be helpful.[4] Those at highest risk of recurrent arrhythmias such as those who have already had a cardiac arrest may benefit from an implantable cardioverter defibrillator – a small device implanted under the skin which can detect dangerous arrhythmias and automatically treat them with a small electric shock.[4]
Periodic paralysis
Periodic paralysis may be improved by taking carbonic anhydrase inhibitors such as acetazolamide.[4]
Epidemiology
Andersen–Tawil syndrome is very rare, and as of 2013 approximately 200 cases had been described in the medical literature.[4] The condition is estimated to affect one person in every 1,000,000.[4]
History
Although a description of the condition had probably been made by Klein in 1963,[4] Andersen–Tawil syndrome is named after Ellen Andersen who described the triad of symptoms in 1971,[13] and Rabi Tawil who made significant contributions to the understanding of the condition in 1994.[14][15]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Veerapandiyan A, Statland JM, Tawil R (2015). "Andersen-Tawil Syndrome". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). GeneReviews. Seattle (WA): University of Washington. PMID 20301441. Archived from the original on 2021-08-26. Retrieved 2021-09-06.
- 1 2 3 4 5 6 7 8 9 "Andersen-Tawil Syndrome - Symptoms, Causes, Treatment | NORD". rarediseases.org. Archived from the original on 7 December 2022. Retrieved 7 May 2023.
- 1 2 3 4 5 6 7 8 "Orphanet: Andersen Tawil syndrome". www.orpha.net. Archived from the original on 14 December 2022. Retrieved 7 May 2023.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 Nguyen HL, Pieper GH, Wilders R (December 2013). "Andersen-Tawil syndrome: clinical and molecular aspects". International Journal of Cardiology. 170 (1): 1–16. doi:10.1016/j.ijcard.2013.10.010. PMID 24383070.
- ↑ "Andersen-Tawil syndrome - About the Disease - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Archived from the original on 28 November 2021. Retrieved 7 May 2023.
- 1 2 3 Tristani-Firouzi M, Etheridge SP (2013). "Chapter 32 – Andersen-Tawil and Timothy Syndromes". In Gussak I, Antzelevitch C (eds.). Electrical diseases of the heart. Volume 1, Basic foundations and primary electrical diseases (2nd ed.). London: Springer. ISBN 978-1-4471-4881-4. OCLC 841465583.
- 1 2 Donaldson MR, Yoon G, Fu YH, Ptacek LJ (2004). "Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity". Annals of Medicine. 36 (Suppl 1): 92–7. doi:10.1080/17431380410032490. PMID 15176430. S2CID 7362563.
- 1 2 Santana, Luis F.; Cheng, Edward P.; Lederer, W. Jonathan (December 2010). "How does the shape of the cardiac action potential control calcium signaling and contraction in the heart?". Journal of Molecular and Cellular Cardiology. 49 (6): 901–903. doi:10.1016/j.yjmcc.2010.09.005. ISSN 1095-8584. PMC 3623268. PMID 20850450.
- 1 2 3 4 Wit, Andrew L. (2018-06-19). "Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias". Pacing and Clinical Electrophysiology. 41 (8): 883–896. doi:10.1111/pace.13419. ISSN 1540-8159. PMID 29920724. S2CID 49310809.
- 1 2 Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, Sansone VA, et al. (April 2018). "Review of the Diagnosis and Treatment of Periodic Paralysis". Muscle & Nerve. 57 (4): 522–530. doi:10.1002/mus.26009. PMC 5867231. PMID 29125635.
- 1 2 Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. (November 2015). "2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC)". Europace. 17 (11): 1601–87. doi:10.1093/europace/euv319. PMID 26318695.
- ↑ Woosley, Raymond L.; Black, Kristin; Heise, C. William; Romero, Klaus (February 2018). "CredibleMeds.org: What does it offer?" (PDF). Trends in Cardiovascular Medicine. 28 (2): 94–99. doi:10.1016/j.tcm.2017.07.010. hdl:10150/627826. ISSN 1873-2615. PMID 28801207. Archived from the original on 2021-03-19. Retrieved 2021-09-06.
- ↑ Andersen ED, Krasilnikoff PA, Overvad H (September 1971). "Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome?". Acta Paediatrica Scandinavica. 60 (5): 559–64. doi:10.1111/j.1651-2227.1971.tb06990.x. PMID 4106724. S2CID 12236896.
- ↑ Tawil R, Ptacek LJ, Pavlakis SG, DeVivo DC, Penn AS, Ozdemir C, Griggs RC (March 1994). "Andersen's syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features". Annals of Neurology. 35 (3): 326–30. doi:10.1002/ana.410350313. PMID 8080508. S2CID 22070260.
- ↑ "Andersen's syndrome (Ellen Damgaard Andersen)". www.whonamedit.com. Archived from the original on 2021-06-24. Retrieved 2019-09-16.
- This article incorporates public domain text from The U.S. National Library of Medicine Archived 2019-02-04 at the Wayback Machine
External links
| Classification | |
|---|---|
| External resources |
|