Atypical hemolytic uremic syndrome
| Atypical hemolytic uremic syndrome | |
|---|---|
| Other names: aHUS | |
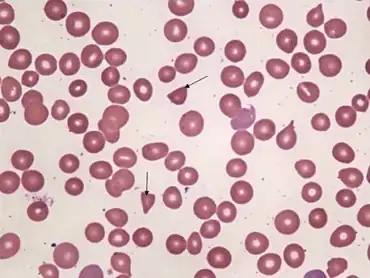 | |
| Peripheral smear -schistocytes arrow | |
Atypical hemolytic uremic syndrome (aHUS) is an extremely rare, life-threatening, progressive disease that frequently has a genetic component. In most cases it can be effectively controlled by interruption of the complement cascade. Particular monoclonal antibodies, discussed later in the article, have proven efficacy in many cases.
AHUS is usually caused by chronic, uncontrolled activation of the complement system,[1][2] a branch of the body’s immune system that destroys and removes foreign particles.[3] The disease affects both children and adults and is characterized by systemic thrombotic microangiopathy (TMA), the formation of blood clots in small blood vessels throughout the body, which can lead to stroke, heart attack, kidney failure, and death.[1][4][5] The complement system activation may be due to mutations in the complement regulatory proteins (factor H, factor I, or membrane cofactor protein),[6][5][7] or is occasionally due to acquired neutralizing autoantibody inhibitors of these complement system components, for example anti–factor H antibodies.[8]: 1933 Prior to availability of eculizumab (Soliris)and ravulizumab(Ultomiris), an estimated 33–40% of patients died or developed end-stage renal disease (ESRD) (despite the use of supportive care, e.g. plasmapheresis) with the first clinical bout of aHUS. Including subsequent relapses, a total of approximately two-thirds (65%) of patients died, required dialysis, or had permanent renal damage within the first year after diagnosis despite plasma exchange or plasma infusion (PE/PI).[7]
Signs and symptoms
Clinical signs and symptoms of complement-mediated TMA can include abdominal pain,[9] confusion,[9] fatigue,[5] edema (swelling),[10] nausea/vomiting[11] and diarrhea.[12] aHUS often presents with malaise and fatigue, as well as microangiopathic anemia.[8]: 1931 However, severe abdominal pain and bloody diarrhea are unusual.[8]: 1931 Laboratory tests may also reveal low levels of platelets (cells in the blood that aid in clotting),[1] elevated lactate dehydrogenase (LDH, a chemical released from damaged cells, and which is therefore a marker of cellular damage),[7] decreased haptoglobin (indicative of the breakdown of red blood cells),[7] anemia (low red blood cell count)/schistocytes (damaged red blood cells),[1][7] elevated creatinine (indicative of kidney dysfunction),[13] and proteinuria (indicative of kidney injury).[14] Patients with aHUS often present with an abrupt onset of systemic signs and symptoms such as acute kidney failure,[1] hypertension (high blood pressure),[5] myocardial infarction (heart attack),[15] stroke,[9] lung complications,[15] pancreatitis (inflammation of the pancreas),[11] liver necrosis (death of liver cells or tissue),[5] encephalopathy (brain dysfunction),[5] seizure,[16] or coma.[17] Failure of neurologic, cardiac, kidney, and gastrointestinal (GI) organs, as well as death, can occur unpredictably at any time, either very quickly or following prolonged symptomatic or asymptomatic disease progression.[1][14][18][19][20] For example, approximately 1 in 6 patients with aHUS initially will present with proteinuria or hematuria without acute kidney failure.[14] Patients who survive the presenting signs and symptoms endure a chronic thrombotic and inflammatory state, which puts many of them at lifelong elevated risk of sudden blood clotting, kidney failure, other severe complications and premature death.[10][21]
Comorbidities
Although many patients experience aHUS as a single disease, comorbidities are common. In one study, 25% (47/191) of patients with no known family history of aHUS were found to have a coexisting disease or condition. Comorbidities in this study included malignant hypertension (30%), TMA with a history of transplant (23%), TMA associated with pregnancy (21%), glomerulopathy (17%), systemic disease such as systemic lupus erythematosus (SLE) or progressive systemic sclerosis (PSS) (6%), and malignancy (1%).[5] The presence of mutations in complement regulatory proteins, or of disease-associated variations in the genes encoding these proteins (i.e., in most patients with comorbid conditions as well as in patients with aHUS as a single disease), suggests that deviations from the normal genetic coding of these factors could result in a genetic predisposition to TMA. Individuals so predisposed could have aHUS episodes precipitated by one of the known disease triggers (e.g., infection, pregnancy, surgery, trauma) as well as by other systemic diseases (e.g., malignant hypertension, SLE, cancer).
Mechanisms
In healthy individuals, complement is used to attack foreign substances, and the complement system is highly regulated to prevent it from damaging healthy tissues and organs.[1][18] However, in most patients with aHUS, it has been demonstrated that chronic, uncontrolled, and excessive activation of complement can result from production of anti-factor H autoantibodies or from genetic mutations in any of several complement regulatory proteins (e.g., factor H, factor HR1 or HR3, membrane cofactor protein, factor I, factor B, complement C3, and thrombomodulin).[18] This results in platelet activation, damage to endothelial cells (cells that line the blood vessels), and white blood cell activation, leading to systemic TMA, which manifests as decreased platelet count, hemolysis (breakdown of red blood cells), damage to multiple organs, and often, death.[14][19][22]
Diagnosis
aHUS is not the only condition that causes systemic TMA, a fact that makes differential diagnosis essential. Historically, the clinical diagnosis of TMA-causing diseases was grouped into a broad category that (in addition to aHUS) included thrombotic thrombocytopenic purpura (TTP) and Shiga-toxin-producing Escherichia coli hemolytic uremic syndrome (STEC-HUS).[19][21] However, it is now understood that although aHUS, STEC-HUS, and TTP have similar clinical presentations, they have distinct causes and specific tests can be conducted to differentiate these diseases. In addition, there are other conditions that can cause TMA as a secondary manifestation; these entities include systemic lupus erythematosus (SLE), malignant hypertension, progressive systemic sclerosis (PSS, also known as scleroderma), the pregnancy-associated HELLP (hemolysis, liver dysfunction, and low platelets) syndrome, and toxic drug reaction (e.g., to cocaine, cyclosporine, or tacrolimus). Nevertheless, aHUS should be suspected in patients presenting with systemic TMA, and appropriate diagnostic work-up should be undertaken.
The neurological and kidney-related signs and symptoms of aHUS overlap with those of TTP.[13][23] However, unlike aHUS, TTP is primarily an autoimmune disorder in which the presence of an inhibitory autoantibody results in severe deficiency of ADAMTS13, an enzyme that cleaves von Willebrand factor (vWf), a large protein involved in blood clotting, into smaller pieces.[22] (TTP also can be a genetic disorder characterized by mutations in the ADAMTS13 gene leading to severe ADAMTS13 deficiency. This congenital cause of ADAMTS13 deficiency is called Upshaw-Schülman syndrome.[24]) A lab test showing ADAMTS13 activity levels of ≤5% is indicative of TTP.[22]
Similarly, the gastrointestinal (GI) signs and symptoms of aHUS overlap with those of STEC-HUS.[12][25][26] Stool samples from patients with diarrhea or other GI symptoms should be tested for STEC and the presence of Shiga-toxin. However, a positive identification of Shiga-toxin, which is required to diagnose STEC-HUS, does not rule out aHUS. Nevertheless, in the appropriate clinical setting, a positive identification of Shiga-toxin makes aHUS very unlikely.[13][26]
aHUS patients report a mean timeline of 29 days for the overall diagnosis process from first noticing symptoms to receiving an aHUS diagnosis. During which time they report their overall health state drops from 88% of patients feeling good to excellent, to 99% feeling good to very poor and a more than halving of their health status index from 3.8 (on a scale of 1 to 5) pre-illness to 1.4 at diagnosis.[27]
Treatment
Plasma exchange/infusion
Although plasma exchange/infusion (PE/PI) is frequently used, there are no controlled trials of its safety or efficacy in aHUS. Even though PE/PI often partially controls some of the hematological manifestations of aHUS in some patients, its effectiveness has not been demonstrated in terms of inducing total disease remission. PE/PI is associated with significant safety risks, including risk of infection, allergic reactions, thrombosis, loss of vascular access, and poor quality of life.[23][28] Importantly, terminal complement activation has been shown to be chronically present on the surface of platelets in patients with aHUS who appear to be clinically well while receiving chronic PE/PI.[10][29]
Guidelines issued by the European Paediatric Study Group for HUS recommend rapid administration of plasma exchange or plasma infusion (PE/PI), intensively administered daily for 5 days and then with reducing frequency.[23] However, the American Society for Apheresis offers a "weak" recommendation for plasma exchange to treat aHUS, due to the "low" or "very low" quality of evidence supporting its use. Although some patients experienced improvements in red blood cell and platelet counts, plasma therapies generally did not result in full remission.[30]
Monoclonal antibody therapy
Eculizumab (Soliris) appears to be useful for atypical hemolytic uremic syndrome (aHUS).[31] In September 2011 the U.S. Food and Drug Administration (FDA) approved it as an orphan drug to treat people with aHUS.[32] This approval was based on two small prospective trials of 17 people and 20 people.[31] In the UK, NICE issued guidance on the use of Eculizumab for treating aHUS, based on five evidence sources, including those used by the FDA[33] No randomised controlled trials were identified. All prospective studies were phase 2, open‑label, non‑randomised, single‑arm studies that included patients with different clinical baseline characteristics. The prospective studies lasted 26 weeks; however, patients were allowed to continue treatment with eculizumab in a long‑term extension study.
Ravulizumab-cwvz (Ultomiris) is a second generation monoclonal antibody for aHUS made by Alexion pharmaceuticals, Inc.[34] The target of ravulizumab-cwvz is the same eculizumab (Soliris) with changes to the structure of the antibody resulting in a longer serum half life and therefore reduced dosing regimen.[35]
Dialysis
Patients with aHUS who have ESRD are generally consigned to lifelong dialysis, which carries a 5-year survival rate of 34–38%,[36][37] with infections accounting for 14% of deaths.[38] These patients also remain at ongoing risk of non-kidney systemic complications of the disease.
Kidney transplantation
Despite its history of use in patients with aHUS, kidney transplantation does not address the continued and uncontrolled complement activation that leads to progressive, systemic TMA. As many as 90% of patients with aHUS and who are not treated with Soliris or Ultomiris, experience TMA in the transplanted organ, leading to transplant failure.[12][39] Patients who have undergone kidney transplantation are still at continued risk of neurological, gastrointestinal, and cardiovascular complications and, importantly, premature mortality. Following kidney transplantation, the ongoing, uncontrolled, chronic complement activation associated with aHUS causes graft loss in 66% of children and 55% of adults, as well as continued inflammatory and TMA insult to other organs.[1][5] Combined liver-kidney transplantation is only available to very few patients, due to the limited supply of solid organs. In addition, there is a substantial near-term risk of mortality, which many physicians and patients consider excessive.[1] In recent years, some transplant centers have begun to administer eculizumab to patients with TMA who receive a kidney transplant. This strategy has been effective in preventing TMA recurrences in these patients.[40]
Terminal complement inhibition
Patients using either eculizumab or ravulizumab for the treatment of aHUS showed improvements in kidney fuction even avoiding dialysis and minimizing death. Markers of disease activity in the blood also had a great improvement. However, the only available evidence has substancial bias and low quality and therefore there should be careful considerations for futures studies in treatment duration, adverse outcomes and risk of disease recurrence associated with this treatment.[41]
Prognosis
Prior to the use of monoclonal antibodies(e.g, Soliris, Ultomiris) patients with aHUS had an extremely poor prognosis. Among those with the most commonly identified aHUS genetic mutation, the proportion of patients experiencing negative outcomes (e.g., need for dialysis, permanent kidney damage, death) within the first year rose to 70%.[7] However, sudden morbidity and mortality could occur regardless of mutational status. aHUS can arise at any age, with more than 40% of cases first reported after 18 years of age.[5] The oldest presentation in one study was at age 83.[5] As noted above, kidney transplantation for aHUS patients with ESRD was rarely considered because of a high incidence of graft loss due to TMA recurrence in the transplanted organ in up to 90% of patients.[12][39] Consequently, most untreated aHUS patients develop ESRD and undergo chronic dialysis, which is associated with significant morbidities and worsened prognosis.[12] Combined liver-kidney transplantation has been attempted in patients with aHUS, although this high-risk procedure has a mortality rate approaching 50%.[42]
Prior to availability and usage of the treatments, quality of life was very poor for patients with aHUS; burdened with fatigue, renal complications, hypertension, neurological impairment, gastrointestinal distress, clotting at the site of venous access, and in worst cases, death.[7] PE/PI is also reported to be associated with significant safety risks and is highly disruptive to patients’ lives due to the requirements for extensive vascular access and frequent administration.[13][43]
Since the approval of eculizumab (Soliris) the prognosis for aHUS patients has improved greatly.[44] Risk of relapse is present after discontinuation of eculizumab treatment and close monitoring is required.[44]
Epidemiology
aHUS can be inherited or acquired, and does not appear to vary by race, gender, or geographic area.[18] As expected with an ultra-rare disease, data on the prevalence of aHUS are extremely limited. A pediatric prevalence of 3.3 cases per million population is documented in one publication of a European hemolytic uremic syndrome (HUS) registry involving 167 pediatric patients.[45]
Society and culture
Naming
Atypical hemolytic uremic syndrome (aHUS) has also been referred to as diarrhea-negative hemolytic-uremic syndrome (D− HUS).[46]: 2170
Research directions
Patient advocacy groups have been helping to determine research priorities.[47]
References
- 1 2 3 4 5 6 7 8 9 Loirat C, Noris M, Fremeaux-Bacchi V (2008). "Complement and the atypical hemolytic uremia syndrome in children". Pediatr Nephrol. 23 (11): 1957–1972. doi:10.1007/s00467-008-0872-4. PMC 6904381. PMID 18594873.
- ↑ Nester, Carla M.; Thomas, Christie P. (8 December 2012). "Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated?". ASH Education Program Book. 2012 (1): 617–625. doi:10.1182/asheducation.v2012.1.617.3798924. ISSN 1520-4391. PMID 23233643.
- ↑ Walport, MJ; Rosen, Fred S.; Walport, Mark J. (2001). "Complement". N Engl J Med. 344 (14): 1058–1066. doi:10.1056/NEJM200104053441406. PMID 11287977.
- ↑ Hosler GA, Cusumano AM, Hutchins GM (2003). "Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases". Arch. Pathol. Lab. Med. 127 (7): 834–9. doi:10.5858/2003-127-834-TTPAHU. PMID 12823037.
- 1 2 3 4 5 6 7 8 9 10 Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. (October 2010). "Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype". Clin J Am Soc Nephrol. 5 (10): 1844–59. doi:10.2215/CJN.02210310. PMC 2974386. PMID 20595690.
- ↑ Tzoumas, Nikolaos; Hallam, Dean; Harris, Claire L.; Lako, Majlinda; Kavanagh, David; Steel, David H.W. (November 2020). "Revisiting the role of factor H in age-related macular degeneration: Insights from complement-mediated renal disease and rare genetic variants". Survey of Ophthalmology. 66 (2): 378–401. doi:10.1016/j.survophthal.2020.10.008. ISSN 0039-6257. PMID 33157112. Archived from the original on 2021-11-20. Retrieved 2021-11-08.
- 1 2 3 4 5 6 7 Caprioli, J; Noris, M; Brioschi, S; Pianetti, G; Castelletti, F; Bettinaglio, P; Mele, C; Bresin, E; Cassis, L; Gamba, S; Porrati, F; Bucchioni, S; Monteferrante, G; Fang, CJ; Liszewski, MK; Kavanagh, D; Atkinson, JP; Remuzzi, G; International Registry of Recurrent Familial HUS/TTP (2006). "Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome". Blood. 108 (4): 1267–1279. doi:10.1182/blood-2005-10-007252. PMC 1895874. PMID 16621965.
- 1 2 3 Hoffman, R; Benz, EJ; Silberstein, LE; Heslop, H; Weitz J; Anastasi, J. (2012). Hematology: Basic Principles and Practice (6th ed.). Elsevier. ISBN 978-1-4377-2928-3.
- 1 2 3 Ohanian, M; Cable, C; Halka, K (2011). "Eculizumab safely reverses neurologic impairment and eliminates the need for dialysis in severe atypical hemolytic uremic syndrome". Clin Pharmacol. 5: 687–696. doi:10.2147/CPAA.S17904. PMC 3262387. PMID 22287852.
- 1 2 3 Stahl, AL; Vaziri-Sani, F; Heinen, S; Kristoffersson, AC; Gydell, KH; Raafat, R; Gutierrez, A; Beringer, O; Zipfel, PF; Karpman, D. (2008). "Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement depositionn on platelets and their activation". Blood. 111 (11): 5307–5315. doi:10.1182/blood-2007-08-106153. PMID 18268093.
- 1 2 Dragon-Durey, M-A; Sethi, SK; Bagga, A; Blanc, C; Blouin, J; Ranchin, B; André, JL; Takagi, N; Cheong, HI; Hari, P.; Le Quintrec, M.; Niaudet, P.; Loirat, C.; Fridman, W. H.; Frémeaux-Bacchi, V. (2010). "Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome". J Am Soc Nephrol. 21 (12): 2180–2187. doi:10.1681/ASN.2010030315. PMC 3014031. PMID 21051740.
- 1 2 3 4 5 Zuber, J; Le Quintrec, Moglie; Sberro-Soussan, Rebecca; Loirat, Chantal; Frémeaux-Bacchi, Véronique; Legendre, Christophe (2011). "New insights into postrenal transplant hemolytic uremic syndrome". Nature Reviews Nephrology. 7 (1): 23–25. doi:10.1038/nrneph.2010.155. PMID 21102542. S2CID 2054556.
- 1 2 3 4 Ariceta, G; Besbas, N; Johnson, S; Karpman, D; Landau, D; Licht, C; Loirat, C; Pecoraro, C; Taylor, CM; Van De Kar, Nicole; Vandewalle, Johan; Zimmerhackl, Lothar B.; European Paediatric Study Group for HUS (2009). "Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome". Pediatr Nephrol. 24 (4): 687–696. doi:10.1007/s00467-008-0964-1. PMID 18800230. S2CID 2967560.
- 1 2 3 4 Sellier-Leclerc, AL; Fremeaux-Bacchi, V; Dragon-Durey, MA; Macher, MA; Niaudet, P; Guest, G; Boudailliez, B; Bouissou, F; Deschenes, G; Gie, S.; Tsimaratos, M.; Fischbach, M.; Morin, D.; Nivet, H.; Alberti, C.; Loirat, C.; French Society of Pediatric Nephrology (2007). "Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome". J Am Soc Nephrol. 18 (8): 2392–2400. doi:10.1681/ASN.2006080811. PMID 17599974.
- 1 2 Sallee, M; Daniel, L; Piercecchi, MD; Jaubert, D; Fremeaux-Bacchi, V; Berland, Y; Burtey, S (2010). "Myocardial infarction is a complication of factor H-associated atypical HUS". Nephrol Dial Transplant. 25 (6): 2028–2032. doi:10.1093/ndt/gfq160. PMID 20305136.
- ↑ Neuhaus, TJ; Calonder, S; Leumann, EP (1997). "Heterogeneity of atypical haemolytic uraemic syndromes". Arch Dis Child. 76 (6): 518–521. doi:10.1136/adc.76.6.518. PMC 1717216. PMID 9245850.
- ↑ Noris, M; Remuzzi, G (2005). "Hemolytic uremic syndrome". J Am Soc Nephrol. 16 (4): 1035–1050. doi:10.1681/ASN.2004100861. PMID 15728781.
- 1 2 3 4 Noris, M; Remuzzi, G (2009). "Atypical hemolytic-uremic syndrome". N Engl J Med. 361 (17): 1676–1687. doi:10.1056/NEJMra0902814. PMID 19846853. S2CID 205115628.
- 1 2 3 Benz, K; Amann, K (2010). "Thrombotic microangiopathy: new insights". Current Opinion in Nephrology and Hypertension. 19 (3): 242–247. doi:10.1097/MNH.0b013e3283378f25. PMID 20186056. S2CID 25429151.
- ↑ Mache, CJ; Acham-Roschitz, B.; Fremeaux-Bacchi, V; Kirschfink, M; Zipfel, PF; Roedl, S; Vester, U; Ring, E (2009). "Complement inhibitor eculizumab in atypical hemolytic uremic syndrome". Clinical Journal of the American Society of Nephrology. 4 (8): 1312–1316. doi:10.2215/CJN.01090209. PMC 2723971. PMID 19556379.
- 1 2 Zipfel, PF; Heinen, S; Skerka, C (2010). "Thrombotic microangiopathies:new insights and new challenges". Current Opinion in Nephrology and Hypertension. 19 (4): 372–378. doi:10.1097/MNH.0b013e32833aff4a. PMID 20539230. S2CID 28419.
- 1 2 3 Tsai, H-M (2010). "Pathophysiology of thrombotic thrombocytopenic purpura". Int J Hematol. 91 (1): 1–19. doi:10.1007/s12185-009-0476-1. PMC 3159000. PMID 20058209.
- 1 2 3 George, JN (2010). "How I treat patients with thrombotic thrombocytopenic purpura". Blood. 116 (20): 4060–4069. doi:10.1182/blood-2010-07-271445. PMID 20686117. S2CID 26844964.
- ↑ Moake, JL (2004). "von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura". Semin Hematol. 41 (1): 4–14. doi:10.1053/j.seminhematol.2003.10.003. PMID 14727254.
- ↑ Fang CJ, Fremeaux-Bacchi V, Liszewski MK, et al. (2008). "Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome". Blood. 111 (2): 624–632. doi:10.1182/blood-2007-04-084533. PMC 2200836. PMID 17914026.
- 1 2 Bitzan, M; Schaefer, Franz; Reymond, Didier (2010). "Treatment of typical (enteropathic) hemolytic uremic syndrome". Semin Thromb Hemostas. 36 (6): 594–610. doi:10.1055/s-0030-1262881. PMID 20865636.
- ↑ "Woodward L, Burke L, Shah K , 2021, Patients' experience and perception of the diagnosis of the rare disease, atypical Hemolytic Uremic Syndrome" (PDF). Archived (PDF) from the original on 2021-09-24. Retrieved 2021-11-08.
- ↑ Michon, B; Moghrabi, A; Winikoff, R; Barrette, S; Bernstein, ML; Champagne, J; David, M; Duval, M; Hume, HA; Robitaille, Nancy; Bélisle, André; Champagne, Martin A. (2007). "Complications of apheresis in children". Transfusion. 47 (10): 1837–1842. doi:10.1111/j.1537-2995.2007.01405.x. PMID 17880609. S2CID 23613105.
- ↑ Licht, C; Pluthero, FG; Li, L; Christensen, H; Habbig, S; Hoppe, B; Geary, DF; Zipfel, PF; Kahr, WH (2009). "Platelet-associated complement factor H in healthy persons and patients with atypical HUS". Blood. 114 (20): 4538–4545. doi:10.1182/blood-2009-03-205096. PMID 19704120.
- ↑ Szczepiorkowski, ZM; Winters, Jeffrey L.; Bandarenko, Nicholas; Kim, Haewon C.; Linenberger, Michael L.; Marques, Marisa B.; Sarode, Ravindra; Schwartz, Joseph; Weinstein, Robert; Shaz, Beth H.; Apheresis Applications Committee of the American Society for Apheresis (2010). "Guidelines on the use of therapeutic apheresis in clinical practice- evidence based approach from the Apheresis Applications Committee of the American Society of Apheresis". J Clin SPheresis. 25 (3): 83–177. doi:10.1002/jca.20240. PMID 20568098. S2CID 7157740.
- 1 2 Keating, GM (December 2013). "Eculizumab: a review of its use in atypical haemolytic uraemic syndrome". Drugs. 73 (18): 2053–66. doi:10.1007/s40265-013-0147-7. PMID 24249647. S2CID 36682579.
- ↑ "FDA approves Soliris for rare pediatric blood disorder: Orphan drug receives second approval for rare disease", FDA, 23 September 2011, archived from the original on 18 January 2017, retrieved 25 June 2015
- ↑ "Eculizumab for treating atypical haemolytic uraemic syndrome | Guidance and guidelines | NICE". nice.org.uk. Archived from the original on 2018-11-26. Retrieved 2021-11-08.
- ↑ "aHUS | Alexion". https://alexion.com. Archived from the original on 2020-08-17. Retrieved 2020-08-19.
{{cite web}}: External link in|website= - ↑ "Ultomiris® (ravulizumab-cwvz) | Alexion". https://alexion.com. Archived from the original on 2020-08-17. Retrieved 2020-08-19.
{{cite web}}: External link in|website= - ↑ Collins, AJ; Foley, Robert N.; Herzog, Charles; Chavers, Blanche M; Gilbertson, David; Ishani, Areef; Kasiske, Bertram L.; Liu, Jiannong; Mau, Lih-Wen; McBean, Marshall (2010). "Excerpts from the US Renal Data System 2009 annual report". Am J Kidney Dis. 55 (1 Suppl 1): S1–S7. doi:10.1053/j.ajkd.2009.10.009. PMC 2829836. PMID 20082919.
- ↑ "European Renal Association-European Dialysis and Transplant Association Registry". ERA-EDTA Registry Annual Report 2009. The Netherlands: Academic Medical Center, Department of Medical Informatics. 2011.
- ↑ Klevens, RM; Edwards, JR; Andrus, ML; Peterson, KD; Dudeck, MA; Horan, TC; NHSN Participants in Outpatient Dialysis Surveillance (2008). "Dialysis surveillance report: National Healthcare Safety Network (NHSN)- data summary for 2006". Semin Dial. 21 (1): 24–28. CiteSeerX 10.1.1.397.9342. doi:10.1111/j.1525-139X.2007.00379.x. PMID 18251954. S2CID 18800012.
- 1 2 Kavanagh, D; Richards, A; Goodship, T; Jalanko, H (2010). "Transplantation in atypical hemolytic uremic syndrome". Semin Thromb Hemost. 36 (6): 653–659. doi:10.1055/s-0030-1262887. PMID 20865642.
- ↑ Nester, C; Stewart, Z; Myers, D; Jetton, J; Nair, R; Reed, A; Thomas, C; Smith, R; Brophy, P (2011). "Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic syndrome". Clin J Am Soc Nephrol. 6 (6): 1488–1494. doi:10.2215/CJN.10181110. PMC 3109948. PMID 21617085.
- ↑ "Interventions for atypical haemolytic uraemic syndrome". Cochrane. Retrieved 2020-11-08.
- ↑ Bresin, E; Daina, E; Noris, M; Castelletti, F; Stefanov, R; Hill, P; Goodship, TH; Remuzzi, G; International Registry of Recurrent Familial HUS/TTP (2006). "Outcome of renal transplantation in patients with non- Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background" (PDF). Clin J Am Soc Nephrol. 1 (1): 88–99. doi:10.2215/CJN.00050505. PMID 17699195. Archived (PDF) from the original on 2018-07-20. Retrieved 2021-11-08.
- ↑ Bouts, A; Monnens, Leo; Davin, Jean-Claude; Struijk, Geertrude; Spanjaard, Lodewijk (2011). "Insufficient protection by Neisseria meningitidis vaccination alone during eculizumab therapy". Pediatr Nephrol. 26 (10): 1919–1920. doi:10.1007/s00467-011-1929-3. PMC 3163808. PMID 21643943.
- 1 2 Menne, Jan; Delmas, Yahsou; Fakhouri, Fadi; Licht, Christoph; Lommelé, Åsa; Minetti, Enrico E.; Provôt, François; Rondeau, Eric; Sheerin, Neil S.; Wang, Jimmy; Weekers, Laurent E. (2019-04-10). "Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study". BMC Nephrology. 20 (1): 125. doi:10.1186/s12882-019-1314-1. ISSN 1471-2369. PMC 6456946. PMID 30971227.
- ↑ Zimmerhackl, LR; Besbas, N; Jungraithmayr, T; Van De Kar, N; Karch, H; Karpman, D; Landau, D; Loirat, C; Proesmans, W; Prüfer, Friederike; Rizzoni, Gianfranco; Taylor, Mark; European Study Group for Haemolytic Uraemic Syndromes Related Disorders (2006). "Epidemiology, clinical presentation, and pathophysiology of atypical and recurrent hemolytic uremic syndrome". Semin Thromb Hemost. 32 (2): 113–120. doi:10.1055/s-2006-939767. PMID 16575686.
- ↑ Kaushansky, K; Lichtman, M; Beutler, E; Kipps, T; Prchal, J; Seligsohn, U. (2010). Williams Hematology (8th ed.). McGraw-Hill. ISBN 978-0071621519.
- ↑ Woodward, Len (2016). "An innovative and collaborative partnership between patients with rare disease and industry-supported registries: the Global aHUS Registry". Orphanet Journal of Rare Diseases. 11 (1): 154. doi:10.1186/s13023-016-0537-5. PMC 5117495. PMID 27871301. Archived from the original on 2017-12-28. Retrieved 2021-11-08.
External links
| Classification | |
|---|---|
| External resources |
|