Autoimmune lymphoproliferative syndrome
| Autoimmune lymphoproliferative syndrome | |
|---|---|
| Other names: Canale-Smith syndrome,[1] | |
Autoimmune lymphoproliferative syndrome (ALPS) is a form of lymphoproliferative disorder (LPDs). It affects lymphocyte apoptosis.[2]
It is a rare genetic disorder of abnormal lymphocyte survival caused by defective Fas mediated apoptosis.[3] Normally, after infectious insult, the immune system down-regulates by increasing Fas expression on activated B and T lymphocytes and Fas-ligand on activated T lymphocytes. Fas and Fas-ligand interact to trigger the caspase cascade, leading to cell apoptosis. Patients with ALPS have a defect in this apoptotic pathway, leading to chronic non-malignant lymphoproliferation, autoimmune disease, and secondary cancers.[4]
Signs and symptoms
All people with ALPS have signs of lymphoproliferation, which makes it the most common clinical manifestation of the disease. The increased proliferation of lymphoid cells can cause the size of lymphoid organs such as the lymph nodes and spleen to increase (lymphadenopathy and splenomegaly, present in respectively over 90% and over 80% of patients). The liver is enlarged (hepatomegaly) in 30–40% of patients.
Autoimmune disease is the second most common clinical manifestation and one that most often requires treatment. The most common autoimmune presentations include autoimmune cytopenias, which can be mild to very severe and intermittent or chronic.[5] These include autoimmune hemolytic anemia, autoimmune neutropenia, and autoimmune thrombocytopenia. Other autoimmune manifestations can be similar to systemic lupus erythematosus (least common, affecting <5% of patients). Manifestations within the nervous system can include autoimmune cerebellar ataxia, Guillain–Barré syndrome, and transverse myelitis. Manifestations in the gastrointestinal system can include atrophic gastritis, and autoimmune hepatitis, esophagitis, colitis, and pancreatitis. Other manifestations can affect the skin (hives), lungs (bronchiolitis obliterans), or kidneys (autoimmune glomerulonephritis and nephrotic syndrome).
Another sign are cancers such as Hodgkin and non-Hodgkin lymphomas, which appear to be increased,[1] possibly due to Epstein–Barr virus-encoded RNA-positivity. Some carcinomas may occur. Unaffected family members with genetic mutations are also at an increased risk of developing cancer.
Genetics
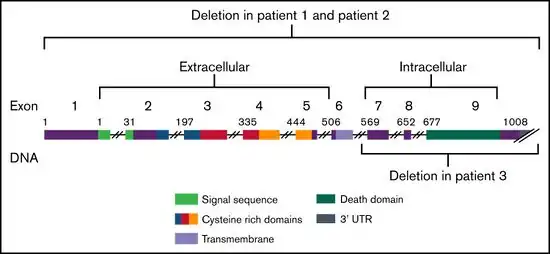
This condition is usually caused by mutations in the FAS gene. Rarely cases due to mutations in other genes including the FAS ligand gene have been reported.[9]
The disease is inherited in an autosomal dominant manner, but it shows incomplete penetrance with up to 40% of people with a FAS mutation not showing symptoms.[10]
Diagnosis
- Elevated peripheral blood Double Negative T cells (DNTs)[11]
- Required for diagnosis
- Immunophenotype: CD3+/CD4-/CD8-/TCRalpha/beta+
- Measured by flow cytometry: Normal values <2.5% total T cells; <1% of total lymphocytes in peripheral blood
- Marked elevations >5% virtually pathognomonic for ALPS
- Mild elevations also found in other autoimmune diseases
- Thought to be cytotoxic T lymphocytes that have lost CD8 expression
- Unknown if driver of disease or epiphenomenon
- May be falsely elevated in setting of lymphopenia or falsely decreased with immunosuppressive treatment
- Biomarkers[12][13]
- Polyclonal hypergammaglobulinemia[14]
- Elevated serum FASL
- Elevated plasma IL-10 and/or IL-18
- Elevated plasma or serum vitamin B12
- Autoantibodies: Non-specific. Can have antibodies to blood cells (DAT, anti-neutrophil, anti-platelet). Also, can have positive ANA, RF, ANCA
- Defective in vitro Fas mediated apoptosis
- Required for diagnosis under old definition. Now can be used to make diagnosis; however, not required to make diagnosis.
- Time and labor-intensive assay.
- T cells from patient and normal control supported in culture for >10 days with mitogen stimulation and IL-2 expansion and then exposed to anti-Fas IgM monoclonal antibody
- ALPS patient T cells: Do not die with anti-Fas monoclonal antibody exposure. Normal T cells from unaffected patient do.
- False negative in somatic Fas variant ALPS and FasL variant ALPS
- Genetic mutations in ALPS causative genes (see below)
Diagnostic algorithm
The old diagnostic criteria for the illness included:[15] Chronic non-malignant lymphoproliferation, elevated peripheral blood DNTs and defective in vitro Fas mediated apoptosis.
The new criteria[16] require chronic non-malignant lymphoproliferation (over six months lymphadenopathy and/or splenomegaly), elevated peripheral blood DNTs. A primary accessory in diagnosis is defective in vitro Fas mediated apoptosis and somatic or germline mutation in ALPS causative gene (FAS, FASL, CASP10).
The secondary accessory in diagnosis are elevated biomarkers (plasma sFASL over 200 pg/ml, plasma IL-10 >20 pg/ml, plasma or serum vitamin B12 >1500 ng/L, Plasma IL-18 >500pg/ml) and immunohistochemical findings on biopsy consistent with ALPS as determined by an experienced hematopathologist. Another sign is autoimmune cytopenias and polyclonal hypergammaglobulinemia and a family history of ALPS or non-malignant lymphoproliferation.
A definitive diagnosis is chronic non-malignant lymphoproliferation and/or elevated peripheral blood DNTs plus one primary accessory criterion. A probable diagnosis is the same but with one secondary accessory criterion.
Classification
2003 nomenclature[15]
- IA - Fas
- IB - Fas ligand
- IIA - Caspase 10
- IIB - Caspase 8
- III - unknown
- IV - Neuroblastoma RAS viral oncogene homolog
Revised nomenclature (2010)[16]
- ALPS-FAS: Fas. Germline FAS mutations. 70% of patients. Autosomal dominant. Dominant negative and haploinsufficient mutations described.[17]
- ALPS-sFAS: Fas. Somatic FAS mutations in DNT compartment.[18] 10% of patients
- ALPS-FASL: Fas ligand. Germline FASL mutations. 3 reported cases
- ALPS-CASP10: Caspase 10. Germline CASP10 mutation. 2% of patients
- ALPS-U: Undefined. 20% of patients
- CEDS: Caspase 8 deficiency state. No longer considered a subtype of ALPS but distinct disorder
- RALD: NRAS, KRAS. Somatic mutations in NRAS and KRAS in lymphocyte compartment. No longer considered a subtype of ALPS but distinct disease
Treatment
Treatment is most commonly directed at autoimmune disease and may be needed to treat bulky lymphoproliferation. First line therapies include corticosteroids (very active but toxic with chronic use), and IVIgG, which are not as effective as in other immune cytopenia syndromes.
Second line therapies include: mycophenolate mofetil (cellcept)[19] which inactivates inosine monophosphate, most studied in clinical trials with responses varying (relapse, resolution, partial response). It does not affect lymphoproliferation or reduce DNTs, with no drug-drug interactions. This treatment is commonly used agent in patients who require chronic treatment based on tolerance and efficacy. It may cause hypogammaglobulinemia (transient) requiring IVIgG replacement.
Sirolimus (rapamycin, rapamune) which is a mTOR (mammalian target of rapamycin) inhibitor[20] can be active in most patients and can in some cases lead to complete or near-complete resolution of autoimmune disease (>90%)[21][22] With this treatment most patients have complete resolution of lymphoproliferation, including lymphadenopathy and splenomegaly (>90%) and have elimination of peripheral blood DNTs. Sirolimus may not be as immune suppressive in normal lymphocytes as other agents. Some patients have had improvement in immune function with transition from cellcept to rapamycin[23] and it has not been reported to cause hypogammaglobulinemia. Hypothetically, Sirolimus may have lower risk of secondary cancers as opposed to other immune suppressants and requires therapeutic drug monitoring. It is the second most commonly used agent in patients that require chronic therapy. It is mostly well tolerated (though side effects include mucositis, diarrhea, hyperlipidemia, delayed wound healing) with drug-drug interactions. It has better activity against autoimmune disease and lymphoproliferation than mycophenolate mofetil and other drugs; however, sirolimus requires therapeutic drug monitoring and can cause mucositis. A risk with any agent in pre-cancerous syndrome as immune suppression can decreased tumor immunosurvellence. Its mTOR inhibitors active against lymphomas, especially EBV+ lymphomas. The Goal serum trough is 5–15 ng/ml and can consider PCP prophylaxis but usually not needed.
Other treatments may include drugs like Fansidar,[24][25] mercaptopurine: More commonly used in Europe. Another is rituximab but this can cause protracted hypogammaglobulinemia[26] and a splenectomy but there is a >30% risk of pneumococcal sepsis even with vaccination and antibiotic prophylaxis[27][28]
References
- 1 2 Straus SE, Jaffe ES, Puck JM; et al. (Jul 2001). "The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis". Blood. 98 (1): 194–200. doi:10.1182/blood.v98.1.194. PMID 11418480.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Fleisher, Thomas A. (2007). "The autoimmune lymphoproliferative syndrome: An experiment of nature involving lymphocyte apoptosis". Immunologic Research. 40 (1): 87–92. doi:10.1007/s12026-007-8001-1. PMID 18193364. S2CID 34565602. Archived from the original on 2023-07-30. Retrieved 2023-07-09.
- ↑ Rao, V. Koneti; Straus, Stephen E. (2006). "Causes and consequences of the autoimmune lymphoproliferative syndrome". Hematology. 11 (1): 15–23. doi:10.1080/10245330500329094. PMID 16522544. S2CID 21782729. Archived from the original on 2023-07-30. Retrieved 2023-07-09.
- ↑ Teachey, David T.; Seif, Alix E.; Grupp, Stephan A. (2010). "Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS)". British Journal of Haematology. 148 (2): 205–16. doi:10.1111/j.1365-2141.2009.07991.x. PMC 2929682. PMID 19930184.
- ↑ Teachey, David T.; Manno, Catherine S.; Axsom, Kelly M.; Andrews, Timothy; Choi, John K.; Greenbaum, Barbara H.; McMann, Joseph M.; Sullivan, Kathleen E.; et al. (2005). "Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS)". Blood. 105 (6): 2443–8. doi:10.1182/blood-2004-09-3542. PMID 15542578.
- ↑ Jevtich, Kathleen; Price, Susan; Similuk, Morgan; Kulm, Elaine; Yan, Jia; Setzer, Michael; Jamal, Leila; Franco, Luis M.; Ghosh, Rajarshi; Walkiewicz, Magdalena; Rao, V. Koneti (12 July 2022). "The contribution of rare copy number variants in FAS toward pathogenesis of autoimmune lymphoproliferative syndrome". Blood Advances. 6 (13): 3974–3978. doi:10.1182/bloodadvances.2021005835. ISSN 2473-9537.
- ↑ Bleesing, Jack JH; Nagaraj, Chinmayee B.; Zhang, Kejian (1993). "Autoimmune Lymphoproliferative Syndrome". GeneReviews®. University of Washington, Seattle.
- ↑ "FAS Fas cell surface death receptor [Homo sapiens (human)] - Gene - NCBI". www.ncbi.nlm.nih.gov. Retrieved 30 July 2023.
- ↑ Magerus-Chatinet A, Stolzenberg MC, Lanzarotti N, Neven B, Daussy C, Picard C, Neveux N, Desai M, Rao M, Ghosh K, Madkaikar M, Fischer A, Rieux-Laucat F (2012) Autoimmune lymphoproliferative syndrome caused by a homozygous null FAS ligand (FASLG) mutation. J Allergy Clin Immunol
- ↑ Autoimmune Lymphoproliferative Syndrome (ALPS) Causes. (2019, April 19). NIH: National Institute of Allergy and Infectious Diseases. https://www.niaid.nih.gov/diseases-conditions/autoimmune-lymphoproliferative-syndrome-causes Archived 2023-06-18 at the Wayback Machine
- ↑ Bleesing, Jack J.H.; Brown, Margaret R.; Novicio, Cynthia; Guarraia, David; Dale, Janet K.; Straus, Stephen E.; Fleisher, Thomas A. (2002). "A Composite Picture of TcRα/β+ CD4−CD8− T Cells (α/β-DNTCs) in Humans with Autoimmune Lymphoproliferative Syndrome". Clinical Immunology. 104 (1): 21–30. doi:10.1006/clim.2002.5225. PMID 12139944.
- ↑ Magerus-Chatinet, Aude; Stolzenberg, Marie-Claude; Loffredo, Maria S.; Neven, Bénédicte; Schaffner, Catherine; Ducrot, Nicolas; Arkwright, Peter D.; Bader-Meunier, Brigitte; et al. (2009). "FAS-L, IL-10, and double-negative CD4−CD8− TCR α/β+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function". Blood. 113 (13): 3027–30. doi:10.1182/blood-2008-09-179630. PMID 19176318. Archived from the original on 2023-07-30. Retrieved 2023-07-09.
- ↑ Caminha, Iusta; Fleisher, Thomas A.; Hornung, Ronald L.; Dale, Janet K.; Niemela, Julie E.; Price, Susan; Davis, Joie; Perkins, Katie; et al. (2010). "Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome". Journal of Allergy and Clinical Immunology. 125 (4): 946–949.e6. doi:10.1016/j.jaci.2009.12.983. PMC 3412519. PMID 20227752.
- ↑ Seif, A. E.; Manno, C. S.; Sheen, C.; Grupp, S. A.; Teachey, D. T. (2010). "Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: A multi-institutional study". Blood. 115 (11): 2142–5. doi:10.1182/blood-2009-08-239525. PMID 20068224.
- 1 2 Sneller, Michael C.; Dale, Janet K.; Straus, Stephen E. (2003). "Autoimmune lymphoproliferative syndrome". Current Opinion in Rheumatology. 15 (4): 417–21. doi:10.1097/00002281-200307000-00008. PMID 12819469. S2CID 21295897. Archived from the original on 2023-07-30. Retrieved 2023-07-09.
- 1 2 Oliveira, J. B.; Bleesing, J. J.; Dianzani, U.; Fleisher, T. A.; Jaffe, E. S.; Lenardo, M. J.; Rieux-Laucat, F.; Siegel, R. M.; et al. (2010). "Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop". Blood. 116 (14): e35–40. doi:10.1182/blood-2010-04-280347. PMC 2953894. PMID 20538792.
- ↑ Kuehn, H. S.; Caminha, I.; Niemela, J. E.; Rao, V. K.; Davis, J.; Fleisher, T. A.; Oliveira, J. B. (2011). "FAS Haploinsufficiency is a Common Disease Mechanism in the Human Autoimmune Lymphoproliferative Syndrome". The Journal of Immunology. 186 (10): 6035–43. doi:10.4049/jimmunol.1100021. PMC 3725553. PMID 21490157.
- ↑ Holzelova, Eliska; Vonarbourg, Cédric; Stolzenberg, Marie-Claude; Arkwright, Peter D.; Selz, Françoise; Prieur, Anne-Marie; Blanche, Stéphane; Bartunkova, Jirina; et al. (2004). "Autoimmune Lymphoproliferative Syndrome with SomaticFasMutations". New England Journal of Medicine. 351 (14): 1409–18. doi:10.1056/NEJMoa040036. PMID 15459302.
- ↑ Koneti Rao, V.; Dugan, Faith; Dale, Janet K.; Davis, Joie; Tretler, Jean; Hurley, John K.; Fleisher, Thomas; Puck, Jennifer; Straus, Stephen E. (2005). "Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome". British Journal of Haematology. 129 (4): 534–8. doi:10.1111/j.1365-2141.2005.05496.x. PMID 15877736.
- ↑ Teachey, D. T.; Obzut, DA; Axsom, K; Choi, JK; Goldsmith, KC; Hall, J; Hulitt, J; Manno, CS; et al. (2006). "Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS)". Blood. 108 (6): 1965–71. doi:10.1182/blood-2006-01-010124. PMC 1895548. PMID 16757690.
- ↑ Teachey, David T.; Greiner, Robert; Seif, Alix; Attiyeh, Edward; Bleesing, Jack; Choi, John; Manno, Catherine; Rappaport, Eric; et al. (2009). "Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome". British Journal of Haematology. 145 (1): 101–6. doi:10.1111/j.1365-2141.2009.07595.x. PMC 2819393. PMID 19208097.
- ↑ Janić, MD; Brasanac, CD; Janković, JS; Dokmanović, BL; Krstovski, RN; Kraguljac Kurtović, JN (2009). "Rapid regression of lymphadenopathy upon rapamycin treatment in a child with autoimmune lymphoproliferative syndrome". Pediatric Blood & Cancer. 53 (6): 1117–9. doi:10.1002/pbc.22151. PMID 19588524.
- ↑ Teachey, David T. (2011). "Autoimmune Lymphoproliferative Syndrome: New Approaches to Diagnosis and Management". Clinical Advances in Hematology & Oncology. 9 (3): 233–5. PMID 21475130. Archived from the original on 2012-04-26.
- ↑ Van Der Werff Ten Bosch, Jutte; Schotte, Peter; Ferster, Alice; Azzi, Nadira; Boehler, Thomas; Laurey, Genevieve; Arola, Mikko; Demanet, Christian; et al. (2002). "Reversion of autoimmune lymphoproliferative syndrome with an antimalarial drug: Preliminary results of a clinical cohort study and molecular observations". British Journal of Haematology. 117 (1): 176–88. doi:10.1046/j.1365-2141.2002.03357.x. PMID 11918552.
- ↑ Rao, V. Koneti; Dowdell, Kennichi C.; Dale, Janet K.; Dugan, Faith; Pesnicak, Lesley; Bi, Lilia L.; Hoffmann, Victoria; Penzak, Scott; et al. (2007). "Pyrimethamine treatment does not ameliorate lymphoproliferation or autoimmune disease in MRL/lpr-/- mice or in patients with autoimmune lymphoproliferative syndrome". American Journal of Hematology. 82 (12): 1049–55. doi:10.1002/ajh.21007. PMID 17674358. S2CID 35312783.
- ↑ Rao, V. Koneti; Price, Susan; Perkins, Katie; Aldridge, Patricia; Tretler, Jean; Davis, Joie; Dale, Janet K.; Gill, Fred; et al. (2009). "Use of rituximab for refractory cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS)". Pediatric Blood & Cancer. 52 (7): 847–52. doi:10.1002/pbc.21965. PMC 2774763. PMID 19214977.
- ↑ Rao, V. K.; Oliveira, J. B. (2011). "How I treat autoimmune lymphoproliferative syndrome". Blood. 118 (22): 5741–51. doi:10.1182/blood-2011-07-325217. PMC 3228494. PMID 21885601.
- ↑ Neven, Bénédicte; Magerus-Chatinet, Aude; Florkin, Benoit; Gobert, Delphine; Lambotte, Olivier; De Somer, Lien; Lanzarotti, Nina; Stolzenberg, Marie-Claude; et al. (2011). "A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation". Blood. 118 (18): 4798–807. doi:10.1182/blood-2011-04-347641. PMID 21885602.
External links
| Classification | |
|---|---|
| External resources |
|