Autosomal dominant GTP cyclohydrolase I deficiency
| Autosomal dominant GTP cyclohydrolase I deficiency | |
|---|---|
| Other names | Autosomal dominant Segawa syndrome (the autosomal recessive form of Segawa syndrome is caused by mutations in a different gene that encodes tyrosine hydroxylase), Dopa-responsive dystonia 5a, Autosomal dominant DYT/PARK-GCH1 (designation in accordance with the Nomenclature of Genetic Movement Disorders maintained by the International Parkinson and Movement Disorder Society[1]) |
Autosomal dominant GTP cyclohydrolase I deficiency (AD-GTPCHD) is a disease caused by dysfunction of GTP cyclohydrolase I, an enzyme that plays an important role in the synthesis of tetrahydrobiopterin, and, as a consequence, of dopamine. This condition is one of the six known causes of tetrahydrobiopterin deficiency[2] and is the most frequently-reported cause of dopa-responsive dystonia.[1]
Symptoms and signs
In more than half the cases, the clinical picture is dominated by postural or action-induced dystonia of one or both lower limbs manifesting as gait difficulties.[2] Dystonia gradually worsens during the day and becomes less pronounced after a period of rest.[2] The fluctuating pattern is highly typical of this disease, especially in the first 30 years, after which this diurnal variation becomes less prominent.[2]
The typical age at onset is during the first decade of life, although an onset in the second decade of life is also common, and in rare cases the disease may present itself in the first 12–18 months of life.[2]
Cause
Autosomal dominant GTP cyclohydrolase I deficiency is caused by mutations in the GCH1 gene that encodes for the enzyme GTP Cyclohydrolase I.[3]
Diagnosis
Diagnosis is complicated because, unlike the majority of BH4 deficiencies, AD-GTPCHD does not present with hyperphenylalaninemia, and is therefore missed during newborn screening.[2] Furthermore, unlike the majority of BH4 deficiencies, this condition does not have a specific pterin pattern.[2] Thus, according to data gathered by the year 2021, the average delay of diagnosis ranged from 8[1] to 10[2] years.
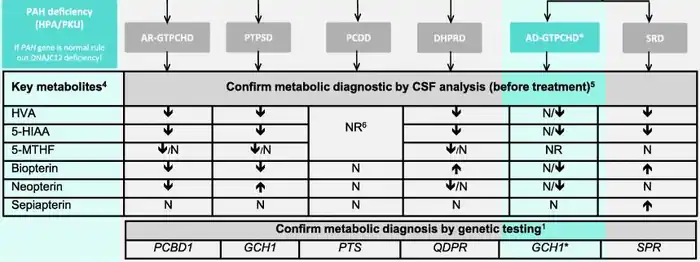
According to a consensus guideline on BH4 deficiency published in 2020, if the presence of AD-GTPCHD is suspected, a genetic assessment should be carried out to look for mutations of the GTPCH1 gene.[2] Genetic assessment requires specialized methods able to detect deletions, because in a significant number of patients with AD-GTPCHD no sequence alterations were found.[2] Furthermore, cerebrospinal fluid analysis of biogenic amines and pterins (Figure 1) should be performed to gain additional clues.[2]
If neither of the first-line tests described above (CSF, genetic tests) are available, a phenylalanine loading test could be performed, which could provide a clue by revealing an increased phenylalanine/tyrosine ratio.[2]
In children whose symptoms are suggestive of dopa-responsive dystonia but for whom neither genetic nor biochemical assessment are available, L-dopa could be briefly prescribed as a trial to see if the patient's condition improves.[2] In case of improvement, the patient would be suspected to have autosomal dominant GTP cyclohydrolase I deficiency, but would still have to undergo genetic and biochemical testing to distinguish their condition from other diseases.[2]
Treatment
Patients are prescribed L-dopa in conjunction with a DC inhibitor such as carbidopa or benserazide.[2] If symptoms persist, dopamine agonists such as pramipexole, bromocriptine, or cabergoline could be considered as a second line of treatment.[2] Anticholinergic drugs or COMT inhibitors could be considered as a third line of treatment.[2]
Epidemiology
As of 2020, there was a lack of precise data on the prevalence and incidence of the disease.[2] One estimate, produced in 2017, puts the prevalence at 2.96 persons in a million people.[2]
See also
References
- 1 2 3 Weissbach A, Pauly MG, Herzog R, Hahn L, Halmans S, Hamami F, Bolte C, Camargos S, Jeon B, Kurian MA, Opladen T, Brüggemann N, Huppertz HJ, König IR, Klein C, Lohmann K (February 2022). "Relationship of Genotype, Phenotype, and Treatment in Dopa-Responsive Dystonia: MDSGene Review". Movement Disorders. 37 (2): 237–252. doi:10.1002/mds.28874. PMID 34908184. S2CID 245260405.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Opladen T, López-Laso E, Cortès-Saladelafont E, Pearson TS, Sivri HS, Yildiz Y, Assmann B, Kurian MA, Leuzzi V, Heales S, Pope S, Porta F, García-Cazorla A, Honzík T, Pons R, Regal L, Goez H, Artuch R, Hoffmann GF, Horvath G, Thöny B, Scholl-Bürgi S, Burlina A, Verbeek MM, Mastrangelo M, Friedman J, Wassenberg T, Jeltsch K, Kulhánek J, Kuseyri Hübschmann O (May 2020). "Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies". Orphanet Journal of Rare Diseases. 15 (1): 126. doi:10.1186/s13023-020-01379-8. PMC 7251883. PMID 32456656.
- ↑ Dayasiri, Kavinda Chandimal; Suraweera, Nayani; Nawarathne, Deepal; Senanayake, U. E.; Dayanath, B. K. T. P.; Jasinge, Eresha; Weerasekara, Kumudu (2019-06-15). "GTP-Cyclohydrolase I deficiency presenting as malignant hyperphenylalaninemia, recurrent hyperthermia and progressive neurological dysfunction in a South Asian child – a case report". BMC Pediatrics. 19 (1): 199. doi:10.1186/s12887-019-1580-x. ISSN 1471-2431. PMC 6570886. PMID 31202265.
External links
- Dystonia, DOPA-responsive, with or without hyperphenylalaninemia- description in the OMIM compendium.