Calpainopathy
| Calpainopathy | |
|---|---|
| Other names: LGMDR1, LMGD2A[1] | |
 | |
| Calpainopathy overview | |
| Specialty | Neurology, neuromuscular medicine |
| Symptoms | proximal muscle weakness, scapular winging |
| Usual onset | 2 - 40 years of age |
| Duration | Long term |
| Types | Pelvifemoral, scapulohumeral, hyperCKemia, autosomal dominant |
| Causes | Genetic (inherited or new mutation) |
| Diagnostic method | Genetic testing |
| Differential diagnosis | Other LGMD2, facioscapulohumeral muscular dystrophy, dystrophinopathy, Metabolic myopathy[2] |
| Management | Physical therapy, bracing, orthopedic surgery |
| Frequency | 1-9/100,000 |
Calpainopathy is the most common type of autosomal recessive limb-girdle muscular dystrophy (LGMD).[3] It preferentially affects the muscles of the hip girdle and shoulder girdle.
No disease modifying pharmaceuticals have been developed as of 2019, although physical therapy, lifestyle modification, and orthopedic surgery can address symptoms.
Signs and symptoms
Disease severity varies greatly, even between family members with identical mutations.[2] Age of onset is highly variable, although symptoms usually appear between 8 and 15 years of age.[4] Patients usually lose the ability to ambulate 10 – 20 years after symptoms appear.[4] Milder forms present with symptoms other than weakness, such as muscle aches, cramps, or exercise intolerance, and people in this group can retain ambulation beyond age 60.[4] Weakness is symmetric, progressive, and proximal (on or close to the torso), usually affecting the hip girdle and shoulder girdle muscles.[2][4] Hip weakness can manifest as a waddling gate.[2] Shoulder weakness can manifest as winged scapulas.[2] Muscle contractures, especially of the Achilles tendon, and scoliosis can also occur.[2]
Heart function and intelligence are generally not affected.[2] Additionally, the muscles of the face, eye, tongue, and neck are spared.[2]
Subtypes
Three subtypes of the autosomal recessive form have been described
- Pelvifemoral (Leyden-Möbius) LGMD: Weakness is first apparent in the pelvic girdle, and later in the shoulder girdle. Onset is early. It is the most frequent subtype.[2]
- Scapulohumeral (Erb) LGMD: Weakness is first apparent the shoulder girdle, and later in the pelvic girdle. Onset is later, and symptoms are milder.[2]
- HyperCKemia: No symptoms, although serum creatine kinase levels are high.[2]
There is a less common, autosomal dominant form, which is milder than the autosomal recessive forms, ranging from no symptoms to wheel chair dependence after age 60.[2]
Genetics
Mutation in the gene CAPN3, which encodes the protein calpain-3 (CAPN3), is the cause of calpainopathy.[2] As of 2019, more than 480 CAPN3 mutations have been reported, some of which can be associated with severe or benign disease course.[4] Usually, the disease follows an autosomal recessive inheritance pattern, requiring both CAPN3 alleles to be mutated for disease to occur.[2] However, there can be CAPN3 mutations that follow an autosomal dominant inheritance pattern.[2]
Pathophysiology


As of 2019, the pathophysiology is largely not understood, although it is increasingly becoming accepted that calcium dysregulation plays a role.[4]
Calpain 3 is unique from other calpain proteases in that it is relatively specific to muscle.[5] Calpain 3 is both a protease and a structural protein.[5] As a protease, it cleaves proteins of the sarcomere and cytoskeleton, designating them to be degraded by proteasomes, a part of muscle remodeling.[5] The structural role of calpain 3 is stabilization of the triad protein complexes.[5] A triad protein complex plays a role converting electrical excitation into calcium release, and it is composed of two calcium channels, the ryanodine receptor (RYR1), and the dihydropyridine receptor (DHPR).[5]
With calpain 3 mutation, proteins typically found at the triad are reduced, including CaMKII (Ca2+/calmodulin-dependent protein kinase II).[5] Decreased CaMKII activity impairs induction of slow oxidative gene expression, which in turn impairs genes involving the mitochondria and lipid metabolism.[5]
Diagnosis
 d) Multi-minicores in a LGMD2A person e) immunohistochemical labelling of N-terminal dystrophin shows decreased or absent expression in a LGMD2A person f) immunohistochemical labelling of dysferlin shows decreased expression in a LGMD2A person
d) Multi-minicores in a LGMD2A person e) immunohistochemical labelling of N-terminal dystrophin shows decreased or absent expression in a LGMD2A person f) immunohistochemical labelling of dysferlin shows decreased expression in a LGMD2A person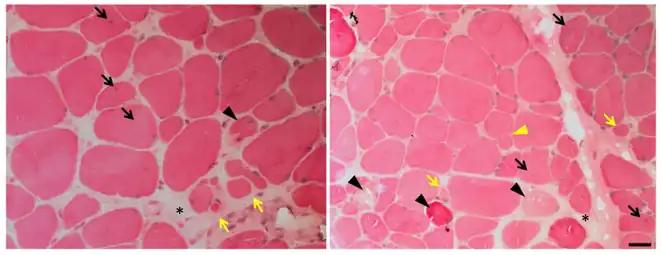 Photomicrograph of muscle affected by calpainopathy. Seen in these views are endomysial fibrosis (black asterisks), central nuclei (black arrows), fiber splitting (yellow triangle), necrosis (black triangles), atrophic fibers (yellow arrows), and increased variation in size and shape.
Photomicrograph of muscle affected by calpainopathy. Seen in these views are endomysial fibrosis (black asterisks), central nuclei (black arrows), fiber splitting (yellow triangle), necrosis (black triangles), atrophic fibers (yellow arrows), and increased variation in size and shape.
Genetic testing is the most definitive test.[2]
If genetic testing is not available, a muscle biopsy with protein immunoanalysis can be used.[2] Biopsy shows general dystrophic features, such as areas of muscle death, variability in muscle size, nuclei in the center of muscle fibers, and disorganized muscle fibers within muscle cells.[4]
Serum creatine kinase, a nonspecific marker of muscle damage, can be elevated early in the disease.[4]
Facioscapulohumeral muscular dystrophy (FSHD) can present similarly, although facial weakness and asymetrical weakness is common in FSHD.
Management
As of 2019, no disease modifying pharmaceuticals are known.[4]
Both strength and aerobic exercise have shown to be beneficial,[4] although strenuous and excessive exercise should be avoided.[2]
Physical therapy can address contractures.[2]
Orthopedic surgery address foot deformities, scoliosis, Achilles tendon contractures, and winged scapula. Winged scapula can be addressed with either scapulopexy or scapulothoracic fusion.[2]
Circumstances to avoid include extremes of body weight, bone fractures, and prolonged immobility.[2]
Epidemiology
Prevalence ranges from 1 to 9 cases per 100,000 people.[4] LGMDR1 represents 30% of all LGMD cases.[4]
History
CAPN3 mutation was the first gene mutation linked to an LGMD.[5]
Research directions
Research is being done to identify the proteins cleaved by calpain-3.[6]
Gene therapy is being studied to replace the function of the calpain-3. Injection of plasmids containing CAPN3 into mouse models resulted in increased levels of calpain-3.[7]
References
- ↑ "Limb-girdle muscular dystrophy: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 1 September 2022. Retrieved 7 September 2022.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Angelini, C; Fanin, M; Adam, MP; Ardinger, HH; Pagon, RA; Wallace, SE; Bean, LJH; Stephens, K; Amemiya, A (1993). "Calpainopathy". PMID 20301490.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Pollitt, C; Anderson, LV; Pogue, R; Davison, K; Pyle, A; Bushby, KM (April 2001). "The phenotype of calpainopathy: diagnosis based on a multidisciplinary approach". Neuromuscular Disorders. 11 (3): 287–96. doi:10.1016/s0960-8966(00)00197-8. PMID 11297944. S2CID 20081475. Archived from the original on 2022-09-20. Retrieved 2022-07-23.
- 1 2 3 4 5 6 7 8 9 10 11 12 Lasa-Elgarresta, J; Mosqueira-Martín, L; Naldaiz-Gastesi, N; Sáenz, A; López de Munain, A; Vallejo-Illarramendi, A (13 September 2019). "Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations". International Journal of Molecular Sciences. 20 (18): 4548. doi:10.3390/ijms20184548. PMC 6770289. PMID 31540302.
- 1 2 3 4 5 6 7 8 Dowling, JJ; Weihl, CC; Spencer, MJ (November 2021). "Molecular and cellular basis of genetically inherited skeletal muscle disorders". Nature Reviews. Molecular Cell Biology. 22 (11): 713–732. doi:10.1038/s41580-021-00389-z. PMID 34257452. S2CID 235822532.
- ↑ Levy, Jennifer (21 January 2020). "Dufour Lab to investigate the biological role of calpain 3 in muscle". Coalition to Cure Calpain 3. Archived from the original on 6 May 2020. Retrieved 6 May 2020.
- ↑ Guha, TK; Pichavant, C; Calos, MP (13 December 2019). "Plasmid-Mediated Gene Therapy in Mouse Models of Limb Girdle Muscular Dystrophy". Molecular Therapy: Methods & Clinical Development. 15: 294–304. doi:10.1016/j.omtm.2019.10.002. PMC 6923511. PMID 31890729.
External links
| Classification |
|---|
| Types | |
|---|---|
| National/International Organizations |
|
| National/International Events |
|
| Clinical trials | |