Combined malonic and methylmalonic aciduria
| Combined malonic and methylmalonic aciduria | |
|---|---|
| Other names: Combined malonic and methylmalonic acidemia | |
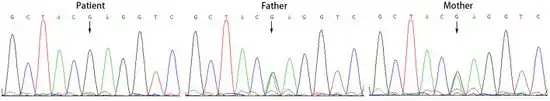 | |
| Sanger sequencing of genomic DNA from individual and relatives with ACSF3 gene (homozygous variant) | |
| Specialty | Medical genetics |
Combined malonic and methylmalonic aciduria (CMAMMA), also called combined malonic and methylmalonic acidemia is an inherited metabolic disease characterized by elevated levels of malonic acid and methylmalonic acid.[1] Some researchers have hypothesized that CMAMMA might be one of the most common forms of methylmalonic acidemia, and possibly one of the most common inborn errors of metabolism.[2] Due to being infrequently diagnosed, it most often goes undetected.[2][3]
Symptoms and signs
The clinical phenotypes of CMAMMA are highly heterogeneous and range from asymptomatic, mild to severe symptoms.[4][5] The underlying pathophysiology is not yet understood.[6] The following symptoms are reported in the literature:
- metabolic acidosis[7][5][8]
- coma[2][6]
- hypoglycemia[2][7][6]
- seizures[2][7][5][8]
- gastrointestinal disease[5][8]
- developmental delay[2][5][8]
- speech delay[1][2][4]
- failure to thrive[2]
- psychiatric disease[2]
- memory problems[2]
- cognitive decline[2]
- encephalopathy[4]
- cardiomyopathy[7][5][8]
- dysmorphic features[5][8]
When the first symptoms appear in childhood, they are more likely to be intermediary metabolic disorders, whereas in adults they are usually neurological symptoms.[2][5]
Causes
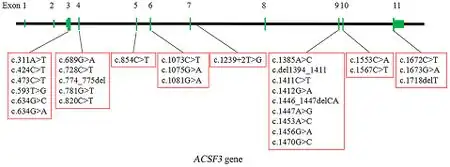
CMAMMA can be divided by causation into two separate inherited disorders: one is a deficiency of the mitochondrial enzyme acyl-CoA synthetase family member 3, encoded by the ACSF3 gene (OMIM#614265); the other disorder is a malonyl-CoA decarboxylase deficiency encoded by the MLYCD gene (OMIM#248360).[1][9]
ACSF3 deficiency
CMAMMA due to ACSF3 is inherited in an autosomal recessive manner. The ACSF3 gene is located on chromosome 16 locus q24.3 and consists of 11 exons and encodes a 576-amino-acid protein.[5][4] CMAMMA can be caused by homozygous or compound heterozygous variants in the ACSF3 gene.[4] Based on minor allele frequency (MAF), a population incidence of ~ 1: 30 000 can be predicted for CMAMMA due to ACSF3.[2]
Malonyl-CoA decarboxylase deficiency
CMAMMA due to malonyl-CoA decarboxylase is also inherited in an autosomal recessive manner. The MLYCD gene is located on chromosome 16 locus q23.3.
Pathophysiology
ACSF3
ACSF3 encodes an acyl-CoA synthetase, which is localized in the mitochondria and has a high specificity for malonic acid and methylmalonic acid. Thus, the synthetase catalyzes the synthesis of malonyl-CoA as well as methylmalonyl-CoA.[9]
Malonic acid
The conversion of malonic acid to malonyl-CoA by acyl-CoA synthetase represents the first step in the mitochondrial fatty acid synthesis (mtFASII) pathway, which plays an important role in the regulation of energy metabolism and which should not be confused with the more familiar fatty acid synthesis that occurs in the cytoplasm.[10][9] The dysfunctional mtFASII pathway leads to an accumulation of malonic acid, which has a competitive inhibitory effect on complex II, and also leads to a deficiency of malonyl-CoA. These deficiencies can be continued to the end-product of the mtFASII pathway, octanoyl-ACP. The consequences of this are diminished oxidative phosphorylation and major alterations in complex lipids, such as increased levels of sphingomyelins and cardiolipins and lower levels of phosphatidylcholines, phosphatidylglycerol and ceramides. Since octanoyl-ACP is the direct precursor in lipoic acid synthesis, this results in diminished lipoylation, since lipoic acid acts as an essential cofactor for several mitochondrial multienzyme complexes, such as pyruvate dehydrogenase complex (PDHC) and α-ketoglutarate dehydrogenase complex (α-KGDHC), among others. This diminished lipoylation also leads to a reduced glycolytic flux.[11][12] To likely compensate for the cell's energy demand, an upregulation of fatty acid β-oxidation and a decreased concentration of certain amino acids that feed anaplerotically into the citrate cycle, such as glutamine (via the fifth site of the citrate cycle), leucine, isoleucine, threonine (all via the sixth site of the citrate cycle) and aspartate (via the 10th site of the citrate cycle), could be detected. In summary, this reduced mitochondrial respiration and glycolytic flux results in impaired mitochondrial flexibility with a large dependence on fatty acid β-oxidation.[6][12]
However, neurons, with the exception of hypothalamic neurons, are not capable of satisfying their large energy demands by degrading fatty acids. It is speculated that an upregulation of β-oxidation also occurs in brain cells due to the hypofunctional mtFASII pathway. The consequence would be an increased risk for hypoxia and oxidative stress, which may contribute to neurological symptoms in the long term.[12]
Methylmalonic acid
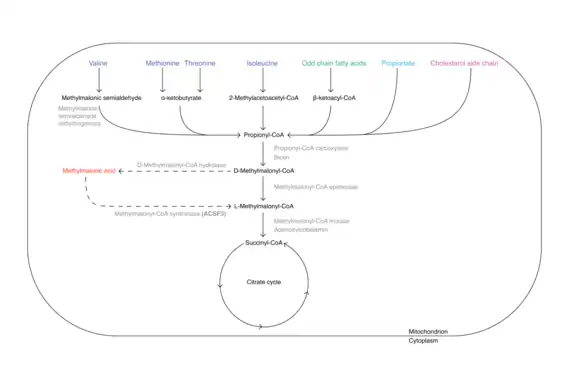
Methylmalonic acid is formed from the essential amino acids valine, methionine, threonine and isoleucine, from odd-chained fatty acids, from propionate and from cholesterol side chain, on the degradation path into the citrate cycle. In this process, the last intermediate methylmalony-CoA can be converted to methymalonic acid by a deacylase before incorporation at the succinyl-CoA site of the citrate cycle. At this point, the acyl-CoA synthease encoded by ACSF3 could convert the methylmalonic acid back to methylmalonyl-CoA and then feed it to the citrate cycle with the help of the methylmalonyl-CoA mutase and its cofactor vitamin B12. However, intracellular esterases are also capable of cleaving the methyl group of methylmalonic acid and generating the parent molecule malonic acid.[13]
Bacterial fermentation in the gut is a quantitatively significant source of propionate, which is a precursor for methylmalonic acid.[14][15] Alongside this, propionic acid is also absorbed through the diet, as it is naturally present in certain foods or is added as a preservative by the food industry, especially in baked goods[16] and dairy products.[17] In addition, methylmalonate is formed during catabolism of thymine.[14][15]
In a study with fibroblasts, increased accumulations of triglycerides, an altered profile of fatty acid chain length and the presence of odd chain fatty acids were detected. A partial degradation due to accumulated methylmalonic acid and the use of propionyl-CoA as a starter unit for fatty acid synthesis is suggested as a possible cause, supported by the observation of a higher expression of CD36, which imports fatty acids into the cell.[6]
In vitro, a connection between free methylmalonic acid and malonic acid to neurotoxicity could be established.[18][13]
Malonyl-CoA decarboxylase deficiency
Malonyl-CoA decarboxylase acts as a catalyst in the conversion of malonyl-CoA to acetyl-CoA and CO2.[19] It is speculated that in MCD deficiency, the excess of mitochondrial malonyl-CoA increases methylmalonic acid levels, as a result of an inhibitory effect on methylmalonyl-CoA mutase.[5][8] In the cytoplasm, malonyl-CoA acts as an inhibitor of the mitochondrial outer membrane enzyme carnitine palmitoyltransferase I (CPT1), which consequently inhibits fatty acid oxidation. The inhibitory effect of cytoplastic malonyl-CoA on CPT1 varies, so it inhibits roughly 100-fold greater in cardiac and skeletal muscles than in the liver.[20]
Diagnosis
Due to a wide range of clinical symptoms and largely slipping through newborn screening programs, CMAMMA is thought to be an under-recognized condition.[1][7]
Newborn screening programs
Because CMAMMA due to ACSF3 does not result in accumulation of methylmalonyl-CoA, malonyl-CoA, or propionyl-CoA, nor are abnormalities seen in the acylcarnitine profile, CMAMMA is not detected by standard blood-based newborn screening programs.[5][2][7]
A special case is the province of Quebec, which, in addition to the blood test, also screens urine on the 21st day after birth with the Quebec Neonatal Blood and Urine Screening Program.[21] This makes Quebec province interesting for CMAMMA research, as it represents the only patient cohort in the world without selection bias.[7] Between 1975 and 2010, an estimated 2 695 000 newborns were thus screened, with 3 detections of CMAMMA due to ACSF3. However, based on this lower detection rate to the predicted rate by heterozygous frequencies, it is likely that not all newborns with this biochemical phenotype were detected by the screening program.[5] A 2019 study then identified as many as 25 CMAMMA due to ACSF3 patients in the province of Quebec.[7] All but one came to clinical attention through the Provincial Neonatal Urine Screening Program, 20 of them directly and 4 after the diagnosis of an older sibling.[7]
Malonic acid to methylmalonic acid ratio
The use of plasma rather than urine is recommended for determining the ratio of malonic acid to methylmalonic acid. Since even with an increase in sensitivity for malonic acid (MA), concentrations in urine samples can be so subtle that they are easily missed. In contrast, if only urinary methylmalonic acid (MAA) is used as the sole matrix, then CMAMMA due to ACSF3 may be misdiagnosed as classic methylmalonic acidemia. Also the calculation of the MA/MAA ratio in urine is not useful, because due to overlapping, healthy individuals cannot be clearly distinguished from CMAMMA affected individuals. Whereas, by calculating the MA/MMA ratio in plasma, a CMAMMA can be clearly distinguished from a classic methylmalonic acidemia. This is true for both, vitamin B12 responders and non-responders in methylmalonic acidemia.[1]
In CMAMMA due to ACSF3, methylamlonic acid levels exceed those of malonic acid. In contrast, in CMAMMA due to malonyl-CoA deficiency, the MMA/MA ratio is less than 1.[8][7]
Genetic testing
Extended carrier screening (ECS) in the course of fertility treatment can also identify carriers of mutations in the ACSF3 gene.[22]
Treatments
Dietary
One approach to reduce the accumulating amount of malonic acid and methylmalonic acid is diet. Here, a diet high in carbohydrate and low in protein has been shown to be best. Changes in malonic acid and methylmalonic acid excretion can be seen as early as 24-36 h after a change in diet.[8]
Bacteria-reducing measures
Another quantitatively significant source of malonic acid and methylmalonic acid, in addition to dietary protein intake, is bacterial fermentation.[14][15] This leads to treatment measures such as the administration of antibiotics and laxatives.
Vitamin B12
Since some forms of methylmalonic acidemia respond to vitamin B12, treatment attempts in CMAMMA due to ACSF3 with vitamin B12 have been made, also in the form of hydroxocobalamin injections, which, however did not lead to any clinical or biochemical effects.[7]
L-Carnitin
One study also mentions treatment with L-carnitine in patients with CMAMMA due to ACSF3, but only retrospectively and without mentioning results.[7]
messenger RNA
Preclinical proof of concept studies in animal models have shown that messenger RNA (mRNA) therapy is also suitable for use in rare metabolic diseases.[23] In this regard, the phase 1/2 study (mRNA-3704 & mRNA-3705) for the treatment of isolated methylmalonic acidemia, which has been ongoing since 2019 by the biotechnology company Moderna, is worth mentioning.[24][25]
Research
In 1984, CMAMMA (due to malonyl-CoA decarboxylase deficiency) was described for the first time in a scientific study.[26][8] Further studies on this form of CMAMMA followed until 1994, when another form of CMAMMA with normal malonyl-CoA decarboxylase activity was discovered.[27][8] In 2011, genetic research through exome sequencing identified the ACSF3 gene as a cause of CMAMMA with normal malonyl-CoA decarboxylase.[2][5] With a study published in 2016, calculation of the MA/MAA ratio in plasma presented a new possibility for rapid, metabolic diagnosis of CMAMMA.[1]
Terminology
The term combined malonic and methylmalonic aciduria with the suffix -uria (from Greek ouron, urine) has become established in the scientific literature in contrast to the other term combined malonic and methylmalonic acedemia with the suffix -emia (from Greek aima, blood). However, in the context of CMAMMA, no clear distinction is made, since malonic acid and methylmalonic acid are elevated in both blood and urine.
See also
References
- 1 2 3 4 5 6 de Sain-van der Velden, Monique G. M.; van der Ham, Maria; Jans, Judith J.; Visser, Gepke; Prinsen, Hubertus C. M. T.; Verhoeven-Duif, Nanda M.; van Gassen, Koen L. I.; van Hasselt, Peter M. (2016), Morava, Eva; Baumgartner, Matthias; Patterson, Marc; Rahman, Shamima (eds.), "A New Approach for Fast Metabolic Diagnostics in CMAMMA", JIMD Reports, Volume 30, Berlin, Heidelberg: Springer Berlin Heidelberg, vol. 30, pp. 15–22, doi:10.1007/8904_2016_531, ISBN 978-3-662-53680-3, PMC 5110436, PMID 26915364
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 NIH Intramural Sequencing Center Group; Sloan, Jennifer L; Johnston, Jennifer J; Manoli, Irini; Chandler, Randy J; Krause, Caitlin; Carrillo-Carrasco, Nuria; Chandrasekaran, Suma D; Sysol, Justin R; O'Brien, Kevin; Hauser, Natalie S (2011). "Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria". Nature Genetics. 43 (9): 883–886. doi:10.1038/ng.908. ISSN 1061-4036. PMC 3163731. PMID 21841779.
- ↑ Sniderman, Lisa C.; Lambert, Marie; Giguère, Robert; Auray-Blais, Christiane; Lemieux, Bernard; Laframboise, Rachel; Rosenblatt, David S.; Treacy, Eileen P. (1999). "Outcome of individuals with low-moderate methylmalonic aciduria detected through a neonatal screening program". The Journal of Pediatrics. 134 (6): 675–680. doi:10.1016/S0022-3476(99)70280-5. PMID 10356133. Archived from the original on 2018-06-11. Retrieved 2023-02-01.
- 1 2 3 4 5 Wang, Ping; Shu, Jianbo; Gu, Chunyu; Yu, Xiaoli; Zheng, Jie; Zhang, Chunhua; Cai, Chunquan (2021). "Combined Malonic and Methylmalonic Aciduria Due to ACSF3 Variants Results in Benign Clinical Course in Three Chinese Patients". Frontiers in Pediatrics. 9: 751895. doi:10.3389/fped.2021.751895. ISSN 2296-2360. PMC 8658908. PMID 34900860.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Alfares, A.; Nunez, L. D.; Al-Thihli, K.; Mitchell, J.; Melancon, S.; Anastasio, N.; Ha, K. C. H.; Majewski, J.; Rosenblatt, D. S.; Braverman, N. (2011). "Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype". Journal of Medical Genetics. 48 (9): 602–605. doi:10.1136/jmedgenet-2011-100230. ISSN 0022-2593. PMID 21785126. S2CID 19352176.
- 1 2 3 4 5 Wehbe, Zeinab; Behringer, Sidney; Alatibi, Khaled; Watkins, David; Rosenblatt, David; Spiekerkoetter, Ute; Tucci, Sara (2019). "The emerging role of the mitochondrial fatty-acid synthase (mtFASII) in the regulation of energy metabolism". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1864 (11): 1629–1643. doi:10.1016/j.bbalip.2019.07.012. PMID 31376476. S2CID 199404906. Archived from the original on 2022-06-19. Retrieved 2023-02-01.
- 1 2 3 4 5 6 7 8 9 10 11 12 Levtova, Alina; Waters, Paula J.; Buhas, Daniela; Lévesque, Sébastien; Auray‐Blais, Christiane; Clarke, Joe T.R.; Laframboise, Rachel; Maranda, Bruno; Mitchell, Grant A.; Brunel‐Guitton, Catherine; Braverman, Nancy E. (2019). "Combined malonic and methylmalonic aciduria due to ACSF3 mutations: Benign clinical course in an unselected cohort". Journal of Inherited Metabolic Disease. 42 (1): 107–116. doi:10.1002/jimd.12032. ISSN 0141-8955. PMID 30740739. S2CID 73436689. Archived from the original on 2022-09-22. Retrieved 2023-02-01.
- 1 2 3 4 5 6 7 8 9 10 11 Gregg, A. R.; Warman, A. W.; Thorburn, D. R.; O'Brien, W. E. (1998). "Combined malonic and methylmalonic aciduria with normal malonyl-coenzyme A decarboxylase activity: A case supporting multiple aetiologies". Journal of Inherited Metabolic Disease. 21 (4): 382–390. doi:10.1023/A:1005302607897. PMID 9700595. S2CID 20212973. Archived from the original on 2022-06-15. Retrieved 2023-02-01.
- 1 2 3 Witkowski, Andrzej; Thweatt, Jennifer; Smith, Stuart (2011). "Mammalian ACSF3 Protein Is a Malonyl-CoA Synthetase That Supplies the Chain Extender Units for Mitochondrial Fatty Acid Synthesis". Journal of Biological Chemistry. 286 (39): 33729–33736. doi:10.1074/jbc.M111.291591. PMC 3190830. PMID 21846720.
- ↑ Bowman, Caitlyn E.; Wolfgang, Michael J. (2019). "Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism". Advances in Biological Regulation. 71: 34–40. doi:10.1016/j.jbior.2018.09.002. PMC 6347522. PMID 30201289.
- ↑ Monteuuis, Geoffray; Suomi, Fumi; Kerätär, Juha M.; Masud, Ali J.; Kastaniotis, Alexander J. (2017-11-06). "A conserved mammalian mitochondrial isoform of acetyl-CoA carboxylase ACC1 provides the malonyl-CoA essential for mitochondrial biogenesis in tandem with ACSF3". Biochemical Journal. 474 (22): 3783–3797. doi:10.1042/bcj20170416. PMID 28986507. Archived from the original on 2023-07-15. Retrieved 2023-02-01.
- 1 2 3 Tucci, Sara (2020). "Brain metabolism and neurological symptoms in combined malonic and methylmalonic aciduria". Orphanet Journal of Rare Diseases. 15 (1): 27. doi:10.1186/s13023-020-1299-7. ISSN 1750-1172. PMC 6977288. PMID 31969167.
- 1 2 McLaughlin, B.A; Nelson, D; Silver, I.A; Erecinska, M; Chesselet, M.-F (1998). "Methylmalonate toxicity in primary neuronal cultures". Neuroscience. 86 (1): 279–290. doi:10.1016/S0306-4522(97)00594-0. PMID 9692761. S2CID 28386770.
- 1 2 3 Thompson, G.N.; Walter, J.H.; Bresson, J.-L.; Ford, G.C.; Lyonnet, S.L.; Chalmers, R.A.; Saudubray, J.-M.; Leonard, J.V.; Halliday, D. (1990). "Sources of propionate in inborn errors of propionate metabolism". Metabolism. 39 (11): 1133–1137. doi:10.1016/0026-0495(90)90084-P. PMID 2233273. Archived from the original on 2022-06-15. Retrieved 2023-02-01.
- 1 2 3 Rosenberg LE (1983). "Disorders of propionate and methylmalonate metabolism". In Stanbury JB, Wyngaarden JB, Frederickson DS (eds.). The metabolic Basis of Inherited Disease (5th ed.). New York. pp. 474–497.
- ↑ Scharinger, Marwa; Kuntz, Marcel; Scharinger, Andreas; Teipel, Jan; Kuballa, Thomas; Walch, Stephan G.; Lachenmeier, Dirk W. (2021-03-03). "Rapid Approach to Determine Propionic and Sorbic Acid Contents in Bread and Bakery Products Using 1H NMR Spectroscopy". Foods. 10 (3): 526. doi:10.3390/foods10030526. ISSN 2304-8158. PMC 7998730. PMID 33802459.
- ↑ Yamamura, T; Okamoto, Y; Okada, G; Takaishi, Y; Takamura, M; Mantani, A; Kurata, A; Otagaki, Y; Yamashita, H; Yamawaki, S (2016). "Association of thalamic hyperactivity with treatment-resistant depression and poor response in early treatment for major depression: a resting-state fMRI study using fractional amplitude of low-frequency fluctuations". Translational Psychiatry. 6 (3): e754. doi:10.1038/tp.2016.18. ISSN 2158-3188. PMC 4872444. PMID 26954981.
- ↑ Kölker, S.; Ahlmeyer, B.; Krieglstein, J.; Hoffmann, G. F. (2000). "Methylmalonic acid induces excitotoxic neuronal damage in vitro". Journal of Inherited Metabolic Disease. 23 (4): 355–358. doi:10.1023/A:1005631230455. PMID 10896293. S2CID 10374239. Archived from the original on 2023-07-15. Retrieved 2023-02-01.
- ↑ Dewit, M; Decoo, I; Verbeek, E; Schot, R; Schoonderwoerd, G; Duran, M; Deklerk, J; Huijmans, J; Lequin, M; Verheijen, F (2006). "Brain abnormalities in a case of malonyl-CoA decarboxylase deficiency". Molecular Genetics and Metabolism. 87 (2): 102–106. doi:10.1016/j.ymgme.2005.09.009. PMID 16275149. Archived from the original on 2022-11-18. Retrieved 2023-02-01.
- ↑ McGarry, J. Denis; Brown, Nicholas F. (1997-02-15). "The Mitochondrial Carnitine Palmitoyltransferase System - From Concept to Molecular Analysis". European Journal of Biochemistry. 244 (1): 1–14. doi:10.1111/j.1432-1033.1997.00001.x. ISSN 0014-2956. PMID 9063439. Archived from the original on 2023-07-15. Retrieved 2023-02-01.
- ↑ "Blood and Urine Screening in Newborns". www.quebec.ca. Archived from the original on 2022-05-23. Retrieved 2022-06-15.
- ↑ Gabriel, Marie Cosette; Rice, Stephanie M.; Sloan, Jennifer L.; Mossayebi, Matthew H.; Venditti, Charles P.; Al‐Kouatly, Huda B. (2021). "Considerations of expanded carrier screening: Lessons learned from combined malonic and methylmalonic aciduria". Molecular Genetics & Genomic Medicine. 9 (4): e1621. doi:10.1002/mgg3.1621. ISSN 2324-9269. PMC 8123733. PMID 33625768.
- ↑ Martini, Paolo G.V.; Guey, Lin T. (2019). "A New Era for Rare Genetic Diseases: Messenger RNA Therapy". Human Gene Therapy. 30 (10): 1180–1189. doi:10.1089/hum.2019.090. ISSN 1043-0342. PMID 31179759. S2CID 182947527. Archived from the original on 2022-02-24. Retrieved 2023-02-01.
- ↑ "A Clinical Trial of a Treatment for Patients with Methylmalonic Acidemia (MMA)". trials.modernatx.com. Archived from the original on 2022-02-18. Retrieved 2022-06-20.
- ↑ "A Clinical Trial of a Treatment for Patients with Methylmalonic Acidemia (MMA)". trials.modernatx.com. Archived from the original on 2022-11-30. Retrieved 2022-06-20.
- ↑ Brown, G. K.; Scholem, R. D.; Bankier, A.; Danks, D. M. (1984). "Malonyl coenzyme a decarboxylase deficiency". Journal of Inherited Metabolic Disease. 7 (1): 21–26. doi:10.1007/BF01805615. ISSN 0141-8955. PMID 6145813. S2CID 33045087. Archived from the original on 2023-07-15. Retrieved 2023-02-01.
- ↑ Ozand, P.T.; Nyhan, W.L.; Al Aqeel, A.; Christodoulou, J. (1994). "Malonic aciduria". Brain and Development. 16: 7–11. doi:10.1016/0387-7604(94)90091-4. PMID 7537025. S2CID 4768844. Archived from the original on 2022-10-22. Retrieved 2023-02-01.
External links
Combined malonic and methylmalonic acidemia Archived 2023-01-27 at the Wayback Machine at Orphanet