D-bifunctional protein deficiency
| D-bifunctional protein deficiency | |
|---|---|
| Other names: 17β-hydroxysteroid dehydrogenase IV deficiency | |
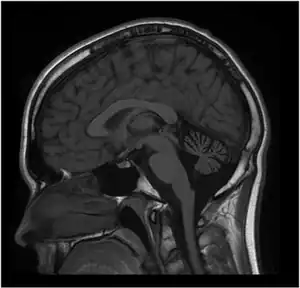 | |
| MRI of the brain. MRI was performed on Person 1 at 12 years of age. T1W1 sagittal image demonstrates prominent cerebellar atrophy involving superior and middle cerebellar folia. Axial images (not shown) revealed normal subcortical and cerebellar white matter. MR spectroscopy was normal (not shown). Repeat MRI imaging at 16 years old was unchanged (not shown). MRI of the brain (Person 2) showed a similar pattern but milder cerebellar atrophy (not shown). | |
| Specialty | Medical genetics |
D-Bifunctional protein deficiency is an autosomal recessive peroxisomal fatty acid oxidation disorder. Peroxisomal disorders are usually caused by a combination of peroxisomal assembly defects or by deficiencies of specific peroxisomal enzymes. The peroxisome is an organelle in the cell similar to the lysosome that functions to detoxify the cell. Peroxisomes contain many different enzymes, such as catalase, and their main function is to neutralize free radicals and detoxify drugs. For this reason peroxisomes are ubiquitous in the liver and kidney. D-BP deficiency is the most severe peroxisomal disorder,[1] often resembling Zellweger syndrome.[2]
Characteristics of the disorder include neonatal hypotonia and seizures, occurring mostly within the first month of life, as well as visual and hearing impairment.[3] Other symptoms include severe craniofacial disfiguration, psychomotor delay, and neuronal migration defects. Most onsets of the disorder begin in the gestational weeks of development and most affected individuals die within the first two years of life.
Classification
DBP deficiency can be divided into three types:[4]
- type I, characterized by a deficiency in both the hydratase and dehydrogenase units of D-BP
- type II, in which only the hydratase unit is non-functional
- type III, with only a deficiency in the dehydrogenase unit
Type I deficient patients showed a large structural modification to the D-BP as a whole. Most of these individuals showed either a deletion or an insertion resulting in a frameshift mutation. Type II and III patients showed small scale changes in the overall structure of D-BP[6]. Amino acid changes in the catalytic domains or those in contact with substrate or cofactors were the main cause of these variations of D-BP deficiency. Other amino acid changes were seen to alter the dimerization of the protein, leading to improper folding. Many mutations have been found in the gene coding for D-BP (HSD17B4) on the q arm two of chromosome five (5q23.1) in Homo sapiens, most notably individuals homozygous for a missense mutation (616S).[4]
Genetic
The D-BP gene (HSD17B4), found on the long arm of chromosome 5, consists of 24 exons and 23 introns and is over 100kb in size. Exons 1-12 code for the SDR domain, 12-21 for the hydratase domain, and 21-24 for the SCP2 domain. Transcription is regulated at 400 basepairs upstream of the transcription start site.[1]
The missense mutation G16S is the most common mutation that leads to D-BP deficiency. In a 2006 study in which 110 patients were tested, 28 suffered from this frameshift mutation. The second most frequent mutation was the missense mutation N457Y which was seen in 13 of the 110 patients. Type I patients showed only deletions, insertions, and nonsense mutations were identified, most leading to shortened polypeptides. Most type II patients show missense mutations in D-BP hydratase unit as well as some in-frame deletions. Type III"individuals commonly show missense mutations in the coding region of the dehydrogenase domain.[4]
D-BP Protein
The D-bifunctional protein is composed of three enzymatic domains: the N-terminal short chain alcohol dehydrogenase reductase (SDR), central hydratase domain, and the C-terminal sterol carrier protein 2 (SDR).[1]
The DBP protein (79kDa) also known as "multifunctional protein 2", "multifunctional enzyme 2", or "D-peroxisomal bifunctional"enzyme", catalyzes the second and third steps of peroxisomal β-oxidation of fatty acids and their derivatives .
A non-functional D-BP protein results in the abnormal accumulation of long chain fatty acids and bile acid intermediates. The D-BP protein contains a peroxisomal targeting signal 1 (PTS1) unit at the C-terminus allowing for its transport into peroxisomes by the PTS1 receptor. Inside the peroxisomes, the D-BP protein is partially cleaved exclusively between the SDR and hydratase"domains.[1]
DBP is a stereospecific enzyme; hydratase domain forms only (R)-hydroxy-acyl-CoA intermediates from trans-2-enoyl-CoAs.[4] D-BP is expressed throughout the entire human body, with the highest mRNA levels in the liver and brain. The hydrogenase and hydratase units of DBP exist as dimers, necessary for correct folding and therefore function of the enzyme.
Chemistry
Enzymatic activity of D-BP fails if the protein cannot effectively bind the cofactor NAD+, as shown in the G16S mutation. Glycine 16 forms a short loop and creates a hole for the adenine ring of NAD+ to enter. Other amino acid side chains alter the shape of this loop due to steric hindrance, and prevent proper NAD+ binding. Other mutations that exist are due to incorrect polypeptide folding. L405 (leucine located at residue 405) located in the substrate binding domain of the hydratase 2 unit, plays an important role in binding CoA ester moiety. One mutation seen in D-BP deficiency patients is caused by a leucine to proline substitution. This breaks the hydrophobic interactions necessary for proper substrate binding with CoA esters.[4]
Diagnosis
The most common clinical observations of patients suffering from D-bifunctional protein deficiency include hypotonia, facial and skull dysmorphism, neonatal seizures, and neuronal demyelination.[5] High levels of branched fatty acids, such as pristinic acid, bile acid intermediates, and other D-BP substrates are seen to exist. Reduced pristinic acid β-oxidation is a common indicator of D-BP deficiency.[1] D-BP can be distinguished from Zellweger Syndrome by normal plasmalogen synthesis. Recent studies in D-BP knockout mice show compensatory upregulation of other peroxisomal enzymes in absence of D-BP such as palmitoyl-CoA oxidase, peroxisomal thiolase, and branched chain acyl-CoA oxidase.[1]
Management
There is no cure for this condition. Management must focus on nutrition and controlling symptoms, while trying to limit any liver disease/damage[6]
References
- 1 2 3 4 5 6 Möller G, van Grunsven EG, Wanders RJ, Adamski J (January 2001). "Molecular basis of D-bifunctional protein deficiency". Mol. Cell. Endocrinol. 171 (1–2): 61–70. doi:10.1016/s0303-7207(00)00388-9. PMID 11165012. S2CID 29712091.
- ↑ Itoh M, Suzuki Y, Akaboshi S, Zhang Z, Miyabara S, Takashima S (March 2000). "Developmental and pathological expression of peroxisomal enzymes: their relationship of D-bifunctional protein deficiency and Zellweger syndrome". Brain Res. 858 (1): 40–7. doi:10.1016/S0006-8993(99)02423-3. PMID 10700594. S2CID 11224543.
- ↑ Buoni S, Zannolli R, Waterham H, Wanders R, Fois A (January 2007). "D-bifunctional protein deficiency associated with drug resistant infantile spasms". Brain Dev. 29 (1): 51–4. doi:10.1016/j.braindev.2006.06.004. PMID 16919904. S2CID 617635.
- 1 2 3 4 5 Ferdinandusse S, Ylianttila MS, Gloerich J, Koski MK, Oostheim W, Waterham HR, Hiltunen JK, Wanders RJ, Glumoff T (January 2006). "Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis". Am. J. Hum. Genet. 78 (1): 112–24. doi:10.1086/498880. PMC 1380208. PMID 16385454.
- ↑ van Grunsven EG, Mooijer PA, Aubourg P, Wanders RJ (August 1999). "Enoyl-CoA hydratase deficiency: identification of a new type of D-bifunctional protein deficiency". Hum. Mol. Genet. 8 (8): 1509–16. doi:10.1093/hmg/8.8.1509. PMID 10400999.
- ↑ "D-bifunctional protein deficiency". NORD (National Organization for Rare Disorders). Archived from the original on 18 August 2021. Retrieved 9 October 2021.
External links
| Classification |
|---|